Org. Synth. 2024, 101, 109-123
DOI: 10.15227/orgsyn.101.0109
Synthesis of 5-(1-Diazo-2-ethoxy-2-oxoethyl) dibenzo[b,d]thiophenium Triflate
Submitted by Brigitte Worbs,
a Sven Timmann,
a Feng Peng,
b Ralph Zhao,
b and Manuel Alcarazo*
1,aChecked by Partha Sarathi Hazra and Kevin Brown
1. Procedure (Note 1)
5-(1-Diazo-2-ethoxy-2-oxoethyl) dibenzo[b,d]thiophenium triflate (2). An oven dried, 500 mL, three-necked, round-bottomed flask (14/23, 29/32, 14/23 ground glass joints), equipped with a Teflon-coated magnetic stir bar (3.5 x 1.0 cm, egg-shaped), is charged with dibenzo[b,d]thiophene 5-oxide (1) (6.0 g, 30 mmol, 1.0 equiv) (Note 2). The flask is fitted with a vacuum adapter (14/23 ground glass joint), while the other two necks are stoppered with new rubber septa. The flask is evacuated (<1 mmHg) and refilled with nitrogen three times. Subsequently, under flow of N2 a low temperature glass thermometer is fitted in the second 14/23 glass joint. Dry dichloromethane (300 mL) (Note 3) is added via cannula through the rubber septum (Figure 1A), and the clear solution is cooled to -78 ℃ (internal temperature) using a cryocooler and an acetone bath (Figure 1B). Triflic acid anhydride (5.0 mL, 30. mmol, 1.0 equiv) is then dropwise added over 30 min by syringe pump, causing an intense red suspension to form (Figure 1C) (Note 4). The reaction mixture is stirred for 1 h at that temperature at which time a solution of ethyl diazoacetate (4.0 mL, 33 mmol, 1.1 equiv) (Note 5) in dry dichloromethane (30 mL) is added to the reaction mixture over 30 min by syringe pump. The clear solution is stirred for an additional hour at -78 ℃ (Figure1D), and then the acetone bath is removed (Figure 1E) and the flask is allowed to warm to ambient temperature over the course of 1 h. The dark orange/red color disappears, and a light brown suspension is formed (Figure 1F).

Figure 1. (A) Solution of 1 in CH2Cl2; (B) Reaction set-up at -78 ℃ prior to the addition of triflic acid anhydride; (C) Orange-red suspension after addition of trific acid anhydride; (D) Clear red-brown solution after addition of ethyl diazoacetate at -78℃; (E) Removal of the Dewar cooling bath; (F) Reaction mixture at room temperature after 1 h (Photos provided by authors)
The reaction mixture is then quenched by addition of water (10 mL) and then poured into a 750 mL separatory funnel, which already contains water (150 mL). To ensure quantitative transfer, the round-bottomed flask is rinsed with dichloromethane (20 mL). The phases are separated (Figure 2A) and the aqueous phase is extracted with dichloromethane (2 x 100 mL) (Figure 2B).
Figure 2. (A) Biphasic mixture after transferring to separatory funnel and shaking; (B) Final extraction of the aqueous phase with CH2Cl2 (photos provided by authors)
The combined organic phases are dried over Na2SO4 (45 g), filtered and transferred to a 1000 mL single-necked, round-bottomed flask. Silica gel (26 g) is added and the resulting slurry is concentrated to dryness, adsorbing the crude product onto silica (Note 6). The product (adsorbed on silica gel) is then loaded onto a packed column prepared from a slurry of silica gel in CH2Cl2 (280 g silica gel, 13.0 x 7.5 cm), and the product layer is covered with a layer of sand (Figure 3A). The column is eluted with 3000 mL of 5% v/v dichloromethane/methanol Figure 3B)(Notes 7, 8, 9, and 10).
Figure 3. (A) Adsorbed product after being loaded onto the column; (B) Column during the elution (photos provided by authors)
The product-containing fractions are combined and concentrated to dryness under reduced pressure on a rotary evaporator (40 ℃, 600 to 30 mmHg) to afford compound (2) as a beige solid (Figure 4A). Then 70 mL of a 1:1 dichloromethane/diethyl ether mixture is added to the solid, and the suspension is sonicated for 10 min (Note 11). The suspension is then cooled to 0 ℃, the solid is filtered under vacuum using a Büchner frit funnel, and washed with a 1:1 dichloromethane/diethyl ether mixture (3 x 10 mL; 0 ℃) (Figure 4B). The white microcrystalline powder is dried under vacuum (20 ℃, 10-3 mmHg) to deliver pure compound 2 (7.31g, 55%) (Figure 4C) (Notes 12, 13, 14, and 15).
Figure 4. (A) Product 2 after evaporation of column chromatography fractions; (B) Compound 2 after the washings; (C) Compound 2 viewed under a microscope (photos provided by authors)
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/content/acs/en/about/governance/committees/chemicalsafety/hazard-assessment.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
dibenzo[b,d]thiophene 5-oxide,
triflic acid anhydride,
ethyl diazoacetate,
dichloromethane,
methanol,
diethyl ether,
sodium sulfate, and silica gel. Diazoacetic esters are potentially explosive and therefore must be handled with caution. They are also toxic and prone to cause development of specific sensitivity. A well-ventilated hood should be used for the entire procedure.
2.
Dibenzo[b,d]thiophene 5-oxide was prepared following a reported method: B. Waldecker, K. Kafuta, M. Alcarazo, Org. Synth.
2019, 96, 258-276. The checkers purchased
dibenzo[b,d]thiophene 5-oxide from Ambeed.
3.
Dichloromethane was purchased from Fischer Chemicals (99.8%) and dried in a MBraun SPS 7 system. The checkers purchased
dichloromethane from Fischer Chemicals (99.8%).
4.
Trifluoromethanesulfonic acid anhydride (97%) was purchased from Fluorochem Ltd. and used as received. The checkers purchased
trifluoromethanesulfonic acid anhydride (97%) from Strem Chemicals, Inc.
5.
Ethyl diazoacetate was purchased from Merck (contains ≥13% wt.
dichloromethane) and used as received. The checkers purchased
ethyl diazoacetate (contains ≥13% wt.
dichloromethane) from Sigma Aldrich.
6. Silica gel 40-63 mesh (grade 60) was purchased from Merck.
7.
Methanol (technical grade) was purchased from Fisher Scientific.
8. Large fractions (25 mL by the checkers and 70 mL by the authors) were collected due to the significant difference in R
f between the products and impurities, which are either very non-polar or stay in the baseline.
9. Fractions 17-39 (author's column) contained pure
2 and were concentrated by rotary evaporation. The product's presence in a fraction is determined by TLC on silica using 10%
methanol in
DCM, showing a R
f of 0.4
Figure 5. Thin layer chromatography (TLC) on silica gel 60 (POLYGRAM SIL G/UV254, polyester sheets) from the fractions of the column chromatography (photo provided by authors)
10. The product front is visible as an orange ring during elution of the column.
11.
Diethyl ether (99.5%) was purchased from Fischer Scientific.
12.
1H NMR
pdf (500 MHz, CD
3CN) δ: 8.29 (d,
J = 8.1 Hz, 2H), 8.23 (dd,
J = 7.9, 1.3 Hz, 2H), 7.91 (td,
J = 7.6, 1.1 Hz, 2H), 7.76 (td,
J = 7.9, 1.2 Hz, 2H), 3.90 (q,
J = 7.1 Hz, 2H), 0.86 (t,
J = 7.2 Hz, 3H);
13C NMR
pdf (126 MHz, CD
3CN) δ: 159.0, 140.2, 135.6, 132.3, 129.5, 128.9, 124.9, 123.4, 64.4, 13.9.
19F NMR
pdf (471 MHz, CD
3CN) δ: -79.29; IR (ATR, cm
-1): 2155.9, 1714.1, 1449.3, 1364.4. 1259.2, 1031.9, 756.7, 639.0, 518.2; mp 148-150 ℃.
13. Differential Scanning Calorimetry was carried out by the authors in a Mettler-Toledo TGA/DSC 3+ system equipped with a SDTA sensor. An aluminum oxide open crucible (70μl) was used with a heating rate of 15 ℃/min. Compound (
2) shows decomposition above 130 ℃ that leads to a significative energy release of ~400 J/g; however, on the basis of the Yoshida correlation, it is predicted not to be explosive or impact sensitive.
2 In addition, no loss of mass is observed when heating a sample of (
2) for 24 h at 50 ℃. For comparison, compound (
4) (see Discussion section) starts decomposing at 120 ℃ with a larger release of energy >600 J/g. On the basis of these results, compound 2 might provide some advantages for multi-gram laboratory scale synthesis.
Figure 6. DSC for 2 shows decomposition on melting above 130 ℃ that leads to significant energy release (400 J/g)
14. The purity of compound
2 (99.4%) was determined by
1H-qNMR
pdf analysis with the relaxation delay set to 30 sec using 43.8 mg (0.098 mmol) of compound
3 and 17.4 mg (0.099 mmol) of dibromomethane (BLD pharm, >99.9%, used as received) as an internal standard.
15. When the reaction was carried out on a 15.0 mmol scale, compound
2 (3.4 g, 51%) was obtained with 98.6% purity using 45 mg (0.1 mmol) of compound
3 and 17.5 mg (0.101 mmol) of dibromomethane (BLD pharm, >99.9%, used as received) as an internal standard.
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
The chemical versatility of diazo compounds, which includes their use as precursors of metal carbenes, reagents for 1,3-dipolar additions, and their native C-nucleophilicity, makes them extremely valuable precursors in organic synthesis.
3 This justifies the enormous attention that has been paid to develop reactions that either allow the convenient installation of this functional group in complex organic architectures,
4 or further broaden the chemical transformations that diazo compounds can accomplish.
5Recently, Suero and coworkers have utilized hypervalent I(III)-reagents
3 and
4 to generate diazomethyl radicals under photochemical conditions and subsequently transfer these radicals to aromatic moieties present in feedstocks and drugs.
6 This new way to introduce diazo moieties in organic skeletons has inspired many researchers, and the number of transformations that employ
3 or
4 has expanded dramatically in the last few years (Scheme 1).
7 Unfortunately, the use of I(III) derivatives is not always straightforward. Often these species undergo strong exothermic decompositions on heating, and some of these compounds are potentially explosive, which limit their use in industrial scale.
8,9 In addition, their strong oxidation potential is often problematic when oxidatively sensitive functionalities are present in the substrate.
Scheme 1. Overview of the reactivity of I(III)-based diazomethyl-transfer reagents
One strategy to develop new reagents that maintain the desired unique reactivity of I(III) reagents but possess increased thermal stability and improved safety profiles relies on the exchange of the benziodoxole moiety with a dibenzothiophenium moiety. Several sulfonium salts, including the well-known Umemoto reagent, make use of this structure/reactivity relationship with their corresponding I(III)-analog.
10Figure 7. Extensively used electrophilic I(III)-based reagents and their sulfonium counterparts
The detailed synthesis of 5-(1-diazo-2-ethoxy-2-oxoethyl) dibenzo[b,d]thiophenium triflate (
2), the sulfonium analogue of
4, is described in this article. Importantly, compound
2 is able to generate diazomethyl radicals under similar, if not identical, conditions to those required by
3 and
4, and therefore shares the same reactivity profile (Scheme 2a).
11 However, differential scanning calorimetry shows that while compound
2 decomposes at 140 ℃ releasing 400 J/g of energy, compound
4 starts decomposing at at lower temperature (120 ℃) with a significantly larger release of energy (>600 J/g). Thus, compound
2 might be considered advantageous for large scale syntheses. In addition, reagent
2 can be purified by column chromatography and kept on the bench in a closed vial for weeks without apparent decomposition, making its handling quite convenient. Finally, it is worth mentioning that the synthetic route herein described can be also employed to prepare sulfonium salts of related structure, such as compounds
5-
7.
11a
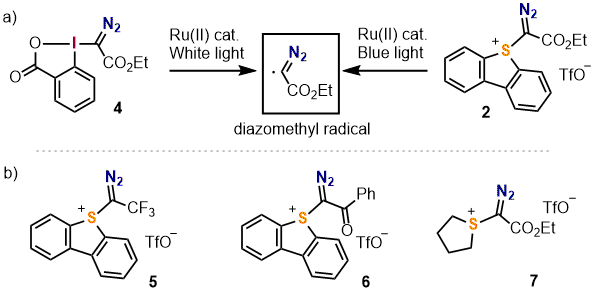
Scheme 2. a) Generation of diazomethyl radicals from 2; b) Additional diazo-substituted sulfonium salts prepared following this route
Appendix
Chemical Abstracts Nomenclature (Registry Number)
Dibenzothiophene, 5-oxide (1013-23-6)
Trifluoromethanesulfonic anhydride (358-23-6)
Ethyl diazoacetate (623-73-4)

|
Brigitte Worbs was born in 1959 in Höxter (Germany), and she started her education in 1976 as a laboratory technical asistant. After completing these studies in 1979, she worked on the synthesis of aromas by Haarmann & Reimer in Holzminden. In 1982 she joined the institute of organic and biomolecular chemistry at the University of Göttingen, and she worked in the groups of Professors Ulrich Schöllkopf (until 1990), Reinhard Brückner (until 1998) and Ulf Diederichsen (until 2021). During that time, she was involved on several projects in the field of organic synthesis. |

|
Sven Timmann obtained his B.Sc. degree in Chemical and Environmental Engineering in 2019 from the University of Applied Science of Lübeck. In 2021 he obtained a M.Sc. degree in Chemistry from the Georg-August University Göttingen under the supervision of Prof. Manuel Alcarazo. He is currently a Ph.D. student in the same group and works on the development of new reactions involving sulfonium salts. |

|
Feng Peng joined the Process Research Department of Merck & Co., Inc. in 2012. His research focuses on using state-of-art organic chemistry to address critical problems in drug development. He received his B. S. degree from Beijing Normal University. He obtained his M.S. under the supervision of Professor Dennis Hall at University of Alberta with a research focus on Boron Chemistry. Feng then moved to New York City, where he obtained Ph.D. in the area of total synthesis (maoecrystal V) with Professor Samuel Danishefsky at Columbia University. |

|
Ralph Zhao is a Principal Scientist in Environmental and Process Safety Engineering (EPSE) group at Merck. His career focus is on process safety field. Currently, he manages all environmental and process safety related issues when Merck transfers small molecule API process from the labs to internal scale-up facilities and external partners. Ralph obtained his B.S. and M. Sc. in Radiation Chemistry from Peking University in China, and Ph.D. in Organic/Polymer chemistry from Virginia Commonwealth University in Richmond, VA under Prof. Raphael Ottenbrite. |

|
Manuel Alcarazo obtained his Master (2002) and his Ph.D. (2005) from the University of Sevilla (Spain). After this, he joined the group of Alois Fürstner as a postdoctoral fellow. He began his independent career at the MPI for Coal Research in Mülheim an der Ruhr (Germany) supported by an ERC Starting Grant, focusing his research on the synthesis and applications of cationic phosphines. In 2015 he was promoted to the rank of full professor at the University of Göttingen (Germany), where he works on the development of new synthetic methodologies in organic chemistry. |

|
Partha Sarathi Hazra was born in Midnapore, West Bengal, India. He received his Bachelor of Science (B.Sc. Hons) in Chemistry from Midnapore College (Autonomous) and Master of Science (M.Sc.) from Indian Institute of Technology (IIT)-Bombay, where he worked with Prof. Debabrata Maiti in C-H activation. Upon completion of his master's study, he moved to Indiana for perusing Ph.D. under the supervision of Prof. M. Kevin Brown at Indiana University-Bloomington in 2022. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved