Org. Synth. 2024, 101, 124-149
DOI: 10.15227/orgsyn.101.0124
Tandem Fragmentation/Olefination for Production of Neopentylene-Tethered 1,6-Enynes from Dimedone
Submitted by Kh Tanvir Ahmed, A. C. Martin, Gabriella O. Wabler, Kristen S. Nerbecki, and Gregory B. Dudley*
1Checked by Hyung Joo Kim and Tehshik Yoon
1. Procedure (Note 1)
A. 5,5-Dimethyl-3-oxocyclohex-1-en-1-yl trifluoromethanesulfonate (1). An oven-dried, 1-L, 3-necked (24/40) round-bottomed flask equipped with a Teflon-coated magnetic stir bar (egg-shaped, 38 x 16 mm), a digital thermometer probe, and nitrogen inlet (Figure 1A) is charged with 5,5-dimethyl-1,3-cyclohexanedione (dimedone, 14.160 g, 0.100 mol, 1.0 equiv) (Note 2). The remaining neck is sealed with a rubber septum, and the flask is evacuated and backfilled with nitrogen in triplicate (Figure 2B). After the addition of methylene chloride (500 mL, 0.2 M) (Note 3) with continuous stirring (600 rpm) at ambient temperature (Note 4), the mixture forms a white suspension (Figure 1C). Pyridine (16.3 mL, 0.200 mol, 2.0 equiv) (Note 5) is added to the reaction mixture. The resulting homogenous solution (Figure 1D) is stirred for 10 min and then placed in a -78 ℃ cooling bath (Notes 6 and 7) for 20 min (Note 8).

Figure 1. General reaction apparatus and addition of reagents; (A) Assembly of the flask, stir bar, nitrogen inlet, and internal thermometer probe; (B) Sealed, nitrogen purged flask with dimedone; (C) addition of dry solvent to flask with dimedone; (D) Clear reaction mixture after the addition of pyridine (photos provided by authors)
Trifluoromethanesulfonic anhydride (triflic anhydride, Tf2O, 20.3 mL, 0.120 mol, 1.2 equiv) (Note 2) is added dropwise (Figure 2A-2D) (Note 9) over 20 min (Notes 10 and 11), followed by stirring in the -78 ℃ cooling bath for 20 min (Note 12).
Figure 2. (A) Triflic anhydride addition and (B-D) progression of color change during the addition process and (E) White cloudy reaction mixture removed from dry ice-acetone bath (photos provided by authors)
The white cloudy reaction mixture is then stirred without external cooling for 2 h (Figure 2E) (Note 13). The reaction is checked using thin layer chromatography (TLC) to ensure completion (Note 14). At the end of the reaction, the rubber septum is removed, and a 250 mL addition funnel is affixed to the flask (Figure 3A). The quenching process is executed by gradually introducing 200 mL of 1.0 M HCl dropwise via the addition funnel over a duration of 15 min (Note 15) (Figure 3A), resulting in a biphasic mixture. The internal temperature reached during the acidification is 18 ℃.
The solution is transferred to a 1-L separatory funnel (Figure 3B). The lower organic phase is removed, and the aqueous layer is extracted with diethyl ether (3 x 100 mL) (Note 16). The organic phases are combined and washed with saturated sodium bicarbonate solution (200 mL) (Note 17) and brine (200 mL) and transferred to a 2-L Erlenmeyer flask. The organic phase is dried over 300 g of sodium sulfate (Note 17) and filtered by vacuum suction through a medium porosity fritted glass funnel (350 mL) into a 2-L evaporating flask. The remaining drying agent left in the filter and the Erlenmeyer flask is washed with an additional 50 mL of diethyl ether (Note 16). The solvent is removed via rotary evaporation (275-35 mmHg, room temperature water bath) (Note 18) providing the crude product as a red oil (Figure 3C) (Note 19). The crude product is purified by column chromatography (Note 20) (Figure 3D and 3E) to afford 26.84 g (99%) of the product as a light-yellow oil (Notes 21, 22, and 23) (Figure 3F).

Figure 3. Workup and purification: (A) quenched reaction mixture, B) in separatory funnel, (C) crude product following rotatory evaporation, D) Purification by column chromatography, E) TLC of column fractions, (F) purified product transferred to a vial (photos provided by authors)
B. 3-Hydroxy-5,5-dimethylcyclohex-1-en-1-yl trifluoromethanesulfonate (2). An oven-dried, 1-L, 3-necked (24/40) round-bottomed flask equipped with a Teflon-coated magnetic stir bar (polygon shaped, 50.8 x 8 mm), a digital thermometer probe, and nitrogen inlet is charged with 5,5-dimethyl-3-oxocyclohex-1-en-1-yl trifluoromethanesulfonate (1) (12.25 g, 45.0 mmol, 1 equiv). The remaining neck is sealed with a rubber septum (Figure 4A). After evacuation and backfilling the flask with nitrogen in triplicate, tetrahydrofuran (180 mL, 0.25M) (Note 24) is added. The reaction mixture is magnetically stirred and cooled to -78 ℃ for 30 min (Notes 6 and 25). Diisobutylaluminium hydride (54 mL, 54.0 mmol, 1.2 equiv) (Figure 4B) (Note 26) is added dropwise over 30 min (Note 27).

Figure 4. (A) Assembly of the flask charged with compound 1 (B) addition of diisobutylaluminium hydride via syringe pump, (C) reaction flask cooled with in ice bath, (D) Appearance of the reaction mixture at room temperature (E) Quenched reaction mixture when warmed to room temperature (photos provided by authors)
The opaque solution is stirred in the -78 ℃ cooling bath for 10 min (Note 28) and the mixture is then kept in an ice bath (Figure 4C) for 10 min (Note 29) followed by stirring for 90 min without external cooling (Note 30), at which point the reaction mixture becomes a homogeneous colorless solution (Figure 4D). The reaction progress is monitored by TLC to confirm consumption of starting material. The reaction mixture is then diluted with diethyl ether (125 mL) (Note 16), cooled to 0 ℃ (Note 31), and quenched by drop-wise addition of distilled water (2.1 mL) over 20 min (Note 32), then 15% NaOH solution (2.1 mL) over 10 min (Note 33), and then distilled water (5.3 mL) over 10 min (Note 34). The mixture is then removed from the ice bath and vigorously stirred for 30 min (Note 35) without external cooling, followed by the addition of 10 g MgSO4 and stirring for an additional 30 min (Note 36).

Figure 5. (A) Filtration through the fritted funnel reaction mixture after quenching and warming to room temperature, (C) crude product as light yellow oil following rotary evaporation (D) Purification by column chromatography (E) TLC of column fractions (F) purified product transferred to a vial (photos provided by authors)
The solution is then filtered by vacuum filtration using a 350 mL ChemGlass medium frit (10-16 μm) filter funnel (Figure 5A). The reaction flask is rinsed with diethyl ether (100 mL), and additional diethyl ether (100 mL) is used to wash the residue on the filter. The filtrate is a transparent colorless solution. The solvent is removed by rotary evaporation (225-35 mmHg, room temperature water bath), providing the crude product as a light-yellow oil (Figure 5B). The crude product is purified (Note 37) by column chromatography (Figure 5C and D) on silica gel to afford 9.39 g (76 %) of the product as a colorless oil (Figure 5E) (Notes 38, 39, and 40).
C. Ethyl (E)-5,5-Dimethyl-oct-2-en-7-ynoate (3). An oven-dried, 500 mL, 3-necked (24/40) round-bottomed flask is equipped with a Teflon-coated magnetic stir bar (egg shaped, 38 x 16 mm), a digital thermometer probe, and nitrogen inlet (Figure 10A). The remaining neck is sealed with a rubber septum. After evacuation and backfilling the flask with nitrogen in three times, tetrahydrofuran (189 mL, 0.1M) (Note 24) and diisopropylamine (6.0 mL, 42.5 mmol, 2.1 equiv) (Note 41) are added by syringes. The reaction is magnetically stirred and placed in a -78 ℃ cooling bath (Notes 6 and 7) for 30 min (Note 42). n-Butyllithium (2.5 M in hexanes, 17.0 mL, 42.5 mmol, 2.1 equiv) (Note 43) is added to the reaction mixture dropwise over 15 min (Note 44).

Figure 6. General Apparatus and Procedure for Fragmentation/Olefination (A) Assembly of the flask, stir bar, nitrogen inlet, and internal thermometer probe (B) addition of n-BuLi to solution of DIPA in THF (C) reaction mixture following warm up to room temperature after the addition of VHAT and triethyl phosphonoacetate (D) Refluxing the reaction using a heating block and condenser (photos provided by authors)
The solution is kept in a -78 ℃ bath for an additional 30 min (Note 45). 3-Hydroxy-5,5-dimethylcyclohex-1-en-1-yl trifluoromethanesulfonate (2) (5.72 g, 20.24 mmol, 1.0 equiv) and triethyl phosphonoacetate (5.09 g, 22.26 mmol, 1.1 equiv) are then added sequentially by syringe (Notes 46 and 47). Following this addition, the colorless solution turns to a light-yellow color. The solution is kept in a -78 ℃ bath for 10 min, then switched to an ice bath for 10 min, and then stirred without external cooling for 50 min (Note 48). When the mixture reaches 18 ℃, the rubber stopper is removed, and a water-cooled condenser is attached. The solution is then heated at reflux for 4 h (Figure 10D) (Note 49). The reaction progress is monitored by TLC (Note 50). After refluxing, the reaction mixture is allowed to cool down to 21 ℃ before quenching.
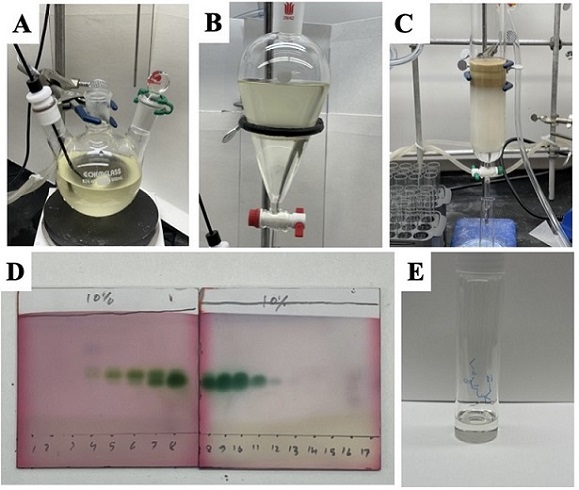
Figure 7. Reaction workup and purification Fragmentation/Olefination (A) Biphasic reaction mixture after the addition of half saturated ammonium chloride (B) quenched reaction mixture in separatory funnel (C) Purification by column chromatography (D) TLC of column fractions (E) purified product transferred to a vial (photos provided by authors)
The reaction is quenched with 80 mL of half-saturated ammonium chloride solution added dropwise (Note 51) and stirred for 5 min. The biphasic mixture (Figure 7A) is then transferred to a 1-L separatory funnel (Figure 7B) (Note 52) and extracted with diethyl ether (3 x 100 mL). The organic layers are combined, washed with water (150 mL), brine (150 mL), and dried over 35 g of anhydrous MgSO4. The dried solution is then filtered through a fluted filter paper (Note 53) into a ground glass funnel attached to a 1L evaporation flask (Note 54). The solvent is removed by rotary evaporation (275-35 mmHg, room temperature water bath), providing the crude product as a light-yellow oil. The crude product is purified by chromatography on silica gel to afford 3.63 g (93%) of the product as a colorless oil (Figure 7E) (Notes 55, 56, 57, and 58).
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/content/acs/en/about/governance/committees/chemicalsafety/hazard-assessment.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
5,5-dimethyl-1,3-cyclohexanedione,
methylene chloride,
pyridine,
trifluoromethanesulfonic anhydride,
hydrochloric acid,
diethyl ether,
sodium bicarbonate,
sodium sulfate,
diisobutylaluminium hydride,
sodium hydroxide,
magnesium sulfate,
tetrahydrofuran,
diisopropylamine,
n-butyllithium,
triethyl phosphonoacetate,
ammonium chloride,
potassium permanganate,
potassium carbonate,
ethyl acetate,
hexane, silica gel,
1,3-benzodioxole,
anisaldehyde,
ethanol, and deuterated chloroform.
2.
5,5-Dimethyl-1,3-cyclohexanedione (
dimedone, 99%) and
trifluoromethanesulfonic anhydride (99.5%) were purchased from TCI Chemicals and Oakwood Chemical, respectively, and used without further purification.
3.
Methylene chloride (HPLC Grade) was purchased from Fischer Scientific and purified via a Pure Process Technology solvent purification system.
4. Ambient temperature in the lab at the start of the reaction was 20 ℃.
5.
Pyridine (99.5%, extra dry) was purchased from Acros Organics as an AcroSeal bottle and used without further purification.
6. The -78 ℃ cooling bath was prepared with acetone and dry ice, and dry ice was periodically added to keep the temperature at -78 ℃.
7. During the entire course of all reactions, the internal temperature is monitored by a digital ChemGlass thermometer (model CG-3497-100) connected to the inserted probe.
8. Internal temperature reached -74 ℃ as indicated by the internal thermometer.
9. All dropwise additions of reagents in all reactions are executed by using New Era Pump System (Model no. 200) unless otherwise mentioned.
10. During
triflic anhydride addition, the solution turned white initially, then light yellow and then changed back to white at the end of
triflic anhydride addition (Figure 2).
11. Maximum internal temperature attained during the addition was -72 ℃.
12. Internal temperature was maintained at or below -74 ℃.
13. Reaction mixture was stirred at 800 rpm. As the temperature increases, the color of the initially white cloudy reaction mixture (Figure 8A) undergoes a series of color transitions, transforming from white to light orange (Figure 8B), then to off-white (Figure 8C), and subsequently to pink (Figure 8D and 8E), culminating in a final shade of red (Figure 8F). The internal temperature eventually reached 15 ℃.
Figure 8. Colorimetric changes of the reaction mixture as the solution temperature warms to 15 ℃ (photos provided by authors)
14. The TLC plates were purchased from Analytical Chemical (TLC Silica Gel 60, Glass Plates 20 x 20cm, cut into 5x2 cm for reaction monitoring and 5 x 5 cm for column fractions screening) and visualized using
potassium permanganate stain, which was prepared as follows: 1.5 g
potassium permanganate, 10 g
potassium carbonate and 1.25 mL 10%
sodium hydroxide was dissolved thoroughly in water. The solution was stored in a wide mouth 100 mL jar with a plastic cap.
15.
Hydrochloric acid (1 M) was prepared by diluting
hydrochloric acid (12.1 M, ACS Grade, purchased from Fisher Scientific) with water.
16.
Diethyl ether (anhydrous, ACS Grade) was purchased from Fisher Chemical and used without further purification.
17.
Sodium bicarbonate (Certified ACS) and
sodium sulfate (anhydrous) were purchased from Fisher Scientific.
18. The temperature of the water bath should be maintained at room temperature to avoid decomposition of the product.
19. The organic phase was transferred to a 1.0 L evaporating flask when the total volume was reduced. Subsequently, 30 mL of additional
diethyl ether was utilized to rinse the 2.0 L flask while transferring the remaining red contents, which reduced to red oil.
20. The crude product was purified using 150 g of Silica Gel (230-400 micro mesh Grade 60), purchased from Fischer Scientific. Silica was dry loaded in a glass column (Diameter: 5 cm) with a fritted disc at the bottom. Sand (30 g) (Fischer Scientific) was added on top of the silica. Solvent (1000 mL of 10%
ethyl acetate:
hexane) was forced through the column to equilibrate the column. The crude material was adsorbed onto silica (30 g), which was dry loaded onto the column followed by a layer of 50 gm sand. After loading the sample, 100 mL of of 10%
ethyl acetate:
hexane was passed through the column in small portions to wet the silica and place the crude material on the top of the silica layer. The crude product was purified using gradient elution (800 mL of 2%
ethyl acetate:
hexanes, 1300 mL of 5%
ethyl acetate:
hexanes) and collected in 50-mL fractions in 25 x 150 mL borosilicate test tubes. The presence of product in the chromatography fractions was identified by TLC analysis on silica, where R
f of
dimedone = 0.1 and R
f of the product (
5,5-dimethyl-3-oxocyclohex-1-en-1-yl trifluoromethane-sulfonate) = 0.55 with 20%
ethyl acetate:
hexanes. Fractions 9-34 were collected (Figure 3E), combined, and concentrated under reduced pressure (
Note 18) to obtain the final product.
21. The product,
5,5-dimethyl-3-oxocyclohex-1-en-1-yl trifluoromethanesulfonate, is colloquially referred to as a vinylogous acyl triflate or VAT. A second run on half-scale provided 13.48 g (99%) of the product. Compound (
1) is obtained as a light-yellow oil, but it darkens upon standing over the course of several days. For prolonged storage (≥1 week), it is recommended to freeze the sample in benzene and repurify immediately before use.
22. The compound exhibited the following physical and spectroscopic data: The boiling point was not determined due to rapid thermal decomposition.
1H NMR
pdf (400 MHz, Chloroform-d) δ: 6.07 (s, 1H), 2.55 (d,
J = 1.3 Hz, 2H), 2.32 (s, 1H), 1.14 (s, 6H);
13C NMR
pdf (100 MHz, Chloroform-d) δ: 197.5, 166.1, 120.1, 118.4, 116.9, 50.7, 42.5, 33.5, 28.1; FTIR (neat) 2965 (w), 2878 (w), 1687 (s), 1648(m), 1424 (s), 1353 (m), 1245 (m), 1207 (vs), 1135 (vs), 1050 (vs), 951 (m), 903 (m), 818 (s), 785 (m), 755 (m) cm
-1.
23. Purity was determined to be >99.5% (average of three runs by qNMR
pdf using 25.9 mg of
1 and 29.5 mg of
1,3-benzodioxole (Thermo Scientific Chemicals, >99.0%, used as received) as the internal standard.
24.
Tetrahydrofuran (HPLC Grade) was purchased from Fischer Scientific and purified via a Pure Process Technology solvent purification system.
25. Internal temperature reached -76 ℃ as indicated by the internal thermometer.
26.
Diisobutylaluminium hydride, a 1.0 M solution in toluene, was purchased from Aldrich Chemistry (Sure-Seal bottle) and used without further purification.
27. During the addition of the
diisobutylaluminium hydride solution, the internal temperature rose to -69 ℃.
28. The internal temperature dropped to -74 ℃.
29. After 10 min of stirring in a mixture of ice and brine, the internal temperature reached -1 ℃.
30. The internal temperature was 20 ℃.
31. The diluted reaction mixture is stirred in a mixture of ice and brine for 15 min. At this point the temperature of the solution is 0 ℃, as indicated by the internal thermometer.
32. Distilled water was obtained through an Aries Filterworks purification system.
33. A solution of
NaOH (15%) was prepared by dissolving 15 g of
NaOH pellets, purchased from Fisher Scientific, in 100 mL of distilled water in an ice bath.
34. The internal temperature remained between 1-2 ℃ during the addition of water and 15%
NaOH. Quenching DIBAL-H reactions can lead to emulsions, but this simple quenching protocol eliminates problematic solids and provides excellent mass recovery of the desired product.
35. A cloudy viscous solution (Figure 4E) forms as the temperature increases, therefore the reaction mixture should be stirred vigorously (1000 rpm). The internal temperature reached 19 ℃ after 30 min.
36.
MgSO4 (anhydrous) was purchased from Fisher Chemicals. The internal temperature reached 22 ℃.
37. The crude product was purified by using column chromatography with 120 gm silica as the stationary phase. The procedure described in
Note 20 was used for column preparation and loading. The crude product is adsorbed onto silica (13 g), which is then loaded onto the packed column. The crude sample is then purified using gradient elution (200 mL of 2%
ethyl acetate:
hexane, 500 mL of 5%
ethyl acetate:
hexane, 500 mL of 10%
ethyl acetate:
hexanes, 500 mL of 15%
ethyl acetate:
hexanes, 1000 mL of 20%
ethyl acetate:
hexanes and collected in 50-mL fractions in 25 x 150 mL borosilicate test tubes. Fractions 19-39 (Figure 5D) were collected, combined, and concentrated under reduced pressure to obtain the final product. The fractions were analyzed by TLC (
Note 14) and visualized using
anisaldehyde stain, which was prepared as follows: To 135 mL of absolute
ethanol was added 5 mL of concentrated sulfuric acid and 1.5 mL of
glacial acetic acid. The resulting solution was cooled to 0 ℃, and 3.7 mL of
p-anisaldehyde was added. The solution is then stirred vigorously to ensure homogeneity. The solution was stored in a wide mouth 100 mL jar with a plastic cap. The R
f of VAT = 0.55 and
3-hydroxy-5,5-dimethylcyclohex-1-en-1-yl trifluoromethanesulfonate = 0.30 in 20%
ethyl acetate:
hexanes.
38.
3-Hydroxy-5,5-dimethylcyclohex-1-en-1-yl is colloquially referred to as vinylogous hemiacetal triflate, VHAT. A second run on 20 mmol scale provided 4.06 g (74%) of the product. The product can be stored in a freezer to avoid decomposition.
39. The compound exhibited the following physical and spectroscopic data: The boiling point was found under reduced pressure (0.03 mmHg) at 60 ℃ (calculated to be 251.6 ℃ at atmospheric pressure).
1H NMR
pdf (500 MHz, Chloroform-d) δ: 5.81 (t,
J = 2.4 Hz, 1H), 4.45 (ddq,
J = 8.7, 5.8, 3.0 Hz, 1H), 2.28 (dt,
J = 17.3, 2.7 Hz, 1H), 2.03 (d,
J = 16.9 Hz, 1H), 1.84 (dd,
J = 13.0, 5.9 Hz, 1H), 1.72 (s, 1H), 1.34 (dd,
J = 13.0, 8.9 Hz, 1H), 1.10 (s, 2H), 0.99 (s, 2H);
13C NMR
pdf (125 MHz, Chloroform-d) δ: 150.0, 120.3, 118.6 (q,
J = 320.2 Hz), 65.8, 44.4, 41.2, 32.4, 30.6, 26.2; FTIR (neat) 3335 (br), 2962(w), 1685 (w), 1415 (s), 1371 (w), 1350 (m), 1245 (m), 1203 (vs), 1138 (vs), 1062 (m), 1013 (m), 953 (m), 932 (m), 903 (m), 882 (m), 856 (m), 827 (m), 783 (w), 759 (m), 607 (vs) cm
-1.
40. Purity was determined to be >99.5% (average of three runs by qNMR
pdf using 51.4 mg of
2 and 59.1 mg of
1,3-Benzodioxole (Thermo Scientific Chemicals, >99.0%, used as received) as the internal standard.
41.
Diisopropylamine (99.5%) was purchased from Acros Organics as an AcroSeal bottle.
42. Internal temperature reached -74 ℃ as indicated by the internal thermometer
43.
n-Butyllithium (2.5 M in
hexanes) was purchased from Sigma-Aldrich in a Sure Seal bottle.
44. During the addition of the
n-BuLi solution, the internal temperature rose to -70 ℃.
45. The internal temperature reached -74 ℃.
46. Compound
2 (97%) and
triethyl phosphonoacetate (98%) were weighed via syringe.
Triethyl phosphonoacetate was purchased from Sigma-Aldrich and used without purification. Both reagents were added dropwise via syringe over a period of 5 min. High purity
triethyl phosphonoacetate produced the best results. The authors report that an older bottle of
triethyl phosphonoacetate resulted in a 10% decrease in yield.
47. During the addition of compound 2 and
triethyl phosphonoacetate, a maximum temperature -71 ℃ was reached. The internal temperature dropped to -74 ℃ after addition was complete.
48. An internal temperature of 18 ℃ was reached.
49. The temperature of the heating mantle was set to 68 ℃, and the 4 h reaction time refers to the time period after the internal temperature reaches 60 ℃. The internal temperature of the solution during the reflux was 64.4 ℃.
50. Analysis by TLC (
Note 14) was performed. The R
f of VHAT = 0.30 and
ethyl (E)-5,5-Dimethyl-oct-2-en-7-ynoate = 0.72 in 20%
ethyl acetate in
hexanes.
51. Half-saturated
ammonium chloride solution was prepared using 19.3 g of
ammonium chloride purchased from Fischer Scientific in 100 mL of distilled water.
52. An additional 50 mL of
diethyl ether was used to rinse the reaction flask into the separatory funnel.
53. Whatman Filter Paper 125 mm circles (Cat No. 1001 125), purchased from Fisher Scientific, were used with a Synthware 24/40 ground glass funnel.
54. The drying agent was rinsed with 100 mL of ethyl ether.
55. The crude product was purified by using column chromatography with 100 gm silica as the stationary phase and 8 gm silica for the sample preparation. The procedure described in Note 20 was used for column preparation and loading. The crude sample was purified using gradient elution (500 mL of 5%
ethyl acetate in
hexanes and 500 mL of 7%
ethyl acetate in
hexanes) and collected in 50-mL fractions in 25 x 150 mm borosilicate test tubes. Fractions 5-12 (Figure 7D) were collected, combined, and concentrated under reduced pressure to obtain the final product. The R
f of VHAT = 0.30 and
ethyl (E)-5,5-Dimethyl-oct-2-en-7-ynoate = 0.72 in 20%
ethyl acetate in
hexanes.
56. A second run on half-scale provided 1.44 g (83%) of the product.
57. The compound exhibited the following physical and spectroscopic data: bp 34 ℃ (0.03 mmHg);
1H NMR
pdf (400 MHz, Chloroform-d) δ: 6.94 (dt,
J = 15.7, 7.9 Hz, 1H), 5.85 (d,
J = 15.5 Hz, 1H), 4.18 (q,
J = 7.1 Hz, 2H), 2.21 (d,
J = 7.9 Hz, 2H), 2.09 - 2.06 (m, 3H), 2.01 (d,
J = 2.4 Hz, 1H), 1.28 (t,
J = 7.1 Hz, 3H), 1.00 (s, 6H);
13C NMR
pdf (100 MHz, Chloroform-d) δ: 166.4, 145.5, 124.0, 81.8, 60.2, 43.6, 34.1, 31.6, 26.7, 14.3; FTIR (neat) 3304 (m), 2962 (m), 1716 (vs), 1653 (m), 1468 (w), 1389 (m), 1368 (m), 1312(m), 1270 (s), 1183 (s), 1155 (m), 1134 (m), 1037 (m), 983 (m) cm
-1.
58. Purity was determined to be 96.7% by qNMR
pdf (average of three runs) using 35.4 mg of
2 and 42.6 mg of 1,2-benzodioxole (Sigma-Aldrich, >99%) as the internal standard.
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Neopentylene-tethered (NPT) 1,6-enynes are valuable building blocks for reaction discovery and target-oriented synthesis. Here they are prepared in three steps from dimedone (5,5-dimethyl-1,3-cyclohexanedone),
2 an inexpensive starting material. The first two steps activate the cyclic structure for ring-opening fragmentation, which is then accomplished in concert with Horner-Wadsworth-Emmons (HWE)-type olefination of the intermediate alkynyl aldehyde ([
I]). Aldehyde [
I] is not stable to the basic reaction conditions and can only be isolated in modest yield (unpublished results), but its slow release by Grob-type anionic fragmentation
3,4 in the presence of the metallated HWE reagent provides for a high yielding olefination. Stereochemistry is established during the olefination event, independent of fragmentation. The resulting tethered alkyne and electron-deficient alkene provide complementary reactivity profiles and the potential for exothermic (cyclo)addition or (cyclo)isomerization reactions.
5
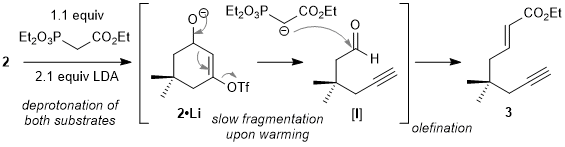
Figure 9. Mechanistic sequence for the base-mediated tandem fragmentation / olefination process
Exploring the chemistry of tethered functional groups is an excellent discovery approach to synthetic methodology. The ideal tether is simple, inert, and effective at orienting complementary functional groups for reactivity. The neopentylene tether is close to ideal in this regard, providing a minimum hydrocarbon framework for Thorpe-Ingold
6 conformational biases. However, access to NPT 1,6-enynes has been challenging. 1,6-Enynes derived from malonates, ethers, or sulfonamides by S
N2-type processes are more readily available, but they bear potentially reactive tether functionality that can interfere with investigations into the desired enyne reactivity profiles. For example, the malonate can conceivably act as a competing ligand or Lewis base in metal-mediated processes, and competing cleavage of allylic ether or sulfonamide bonds can also be envisioned. There are thus clear advantages to reaction discovery efforts using NPT 1,6-enyne substrates.
NPT 1,6-enynes are also of strategic value for natural product synthesis and for accessing chemical structure space that is largely missing from synthetic pharmaceutical libraries. The neopentylene subunit maps conveniently onto a number of classic sesquiterpene targets for total synthesis (Figure 10). For example, the tandem fragmentation / olefination approach to NPT 1,6-enynes presented here
2, formally shortens previous syntheses of hirsutene
7 and illudol.
8 NPT 1,6-enynes would likewise enhance the scope of diversity-oriented synthesis programs that leverage the broad reactivity profile of 1,6-enynes.
9 Better access to NPT 1,6-enynes advances target- and diversity-oriented synthesis and creates opportunities in drug discovery.
Figure 10. Examples of sesquiterpene natural products bearing a neopentylene ring fusion (gem-dimethylcyclopentane)
A set of representative examples of NPT 1,6-enynes prepared by this method is provided in Figure 11. Most were prepared using standard HWE reagents.
2,10 Other carbanionic nucleophiles can intercept aldehyde [
I], provided that the nucleophiles are inert to triflate
2 and can withstand the mild heating used to promote fragmentation. Traditional Wittig-type phosphorus ylide reagents did not prove to be competent partners in preliminary experiments. Vinylpyridines
1111 and
1212 were prepared by Knoevenagel-type condensations using simple pyridylacetic acid derivatives; no phosphonate activating group was necessary. Vinyl sulfide
1413 realizes the possibility of making electron-rich alkenes, which has not been systematically explored in this methodology.
Figure 11. Selected examples of NPT 1,6-enynes prepared from dimedone (and 1,6 enyne 5, prepared from 2-methyl-1,3-cyclohexanedione) by this tandem fragmentation / olefination process
NPT dienyne
10 highlights the utility of NPT substrates in reaction discovery.
14 In an initial exploratory experiment, this electron-deficient dienyne underwent an oxidative cycloisomerization process in the presence of 10 mol% Wilkinson's catalyst at 130 ℃, whereas the analogous malonate-, sulfonamide-, and ether-tethered substrates did not. This process features intramolecular [4+2] cycloaddition of a dienoate with an unactivated alkyne. Such transformations are often problematic, as discussed elsewhere.
15 Further development from this initial reaction discovery revealed that various indanes can be prepared at room temperature using 1 mol% of a cationic Rh dimer catalyst, whereas dienynes bearing heteroatoms in the tether require higher catalyst loading (Figure 12). From competition experiments we tentatively concluded that electronegative heteroatoms in the tether inductively deactivate the substrate. NPT substrates were instrumental in this discovery and development, whereas a methodology that relied on 1,6-enynes bearing heteroatoms in the tether might have overlooked this process.
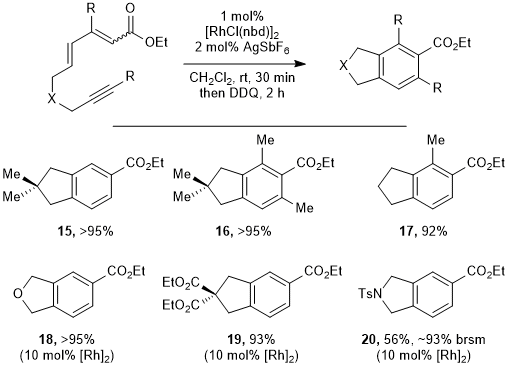
Figure 12. A Rh-catalyzed dienyne oxidative cycloisomerization, in which NPT dienynes outperform those with heteroatoms in the tether
Last but not least, NPT 1,6-enynes are the missing link between enyne cycloisomerization methodologies and their applications to the synthesis of the sesquiterpene natural products bearing neopentylene ring fusions. As noted above, neopentylene subunits map conveniently onto a number of natural product targets. Indane
16 (Figure 12) was a late-stage intermediate in the first synthesis of alcyopterosin A,
16 prepared previously in 9 linear steps from isophorone, and now in 4 steps from dimedone.
17 We reported syntheses of alcyopterosin A
17 and illudinine
10,11 in 6 and 7 linear steps from dimedone, respectively (Figure 13) - in each case leveraging NPT enyne substrates to lower the step-count and increase the overall yield compared to previous syntheses of the target compounds.
Figure 13. Strategic applications of NPT 1,6-enynes in synthesis
In summary, NPT 1,6-enynes are readily available in three steps from dimedone. NPT 1,6-enynes are valuable building blocks for target-oriented synthesis, and they approximate the ideal substrate for 1,6-enyne annulation and/or cycloisomerization reaction discovery, which continues to be a fruitful strategy for innovation in reaction methodology.
Appendix
Chemical Abstracts Nomenclature (Registry Number)
5,5-Dimethyl-3-oxocyclohex-1-en-1-yl trifluoromethanesulfonate: Methanesulfonic acid, 1,1,1-trifluoro-, 5,5-dimethyl-3-oxo-1-cyclohexen-1-yl ester; (1) (76881-19-1)
3-Hydroxy-5,5-dimethylcyclohex-1-en-1-yl trifluoromethanesulfonate: Methanesulfonic acid, 1,1,1-trifluoro-, 3-hydroxy-5,5-dimethyl-1-cyclohexen-1-yl ester; (2) (109459-33-8)
(E)-4,4-Dimethyl-1-(phenylsulfonyl)-1-hepten-6-yne: 2-Octen-7-ynoic acid, 5,5-dimethyl-, ethyl ester, (2E)-; (517893-66-2)

|
Kh Tanvir Ahmed was born in Dhaka, Bangladesh and received B.Pharm. and M.Pharm from the University of Dhaka. He completed M.S. in Medicinal Chemistry from Duquesne University in 2020. He is currently pursuing Ph.D. in Chemistry at the West Virginia University under the supervision of Professor Gregory B. Dudley. His research focuses on regioselective cyclotrimerization, total synthesis of illudalane type sesquiterpenes and analog design from illudalic acid to target protein tyrosine phosphatase enzymes. |

|
A. C. Martin was born and raised in Brownsville, Pennsylvania, a suburb of Pittsburgh. She received her B.S. degree in chemistry in 2017 from the University of Pittsburgh, and she completed her Ph.D. degree under the co-supervision of Professor Brian Popp and Professor Gregory Dudley in 2022. Her research focused on metal catalyzed cyclotrimerization of 1,6-enynes. |

|
Gabriella O. Wabler was born and raised in Dayton, Ohio. She graduated West Virginia University with a B.S. degree in biochemistry in 2020, after doing undergraduate research in the Dudley lab. |

|
Kristen S. Nerbecki was born and raised in Pittston, Pennsylvania. She received her B.S. in Chemistry in the Spring of 2019 from Bridgewater College, and she completed her M.S. degree under Professor Gregory Dudley in 2022. Her research focused on tandem addition/fragmentation reactions with various vinylogous acyl sulfonates. |

|
Gregory B. Dudley is the Eberly Professor and Chair at WVU Chemistry. He received a B.A. from FSU in 1995 and a Ph.D. in 2000 under the direction of Rick Danheiser at MIT, then completed an NIH Postdoctoral Fellowship with Samuel Danishefsky at the Sloan-Kettering Institute for Cancer Research. He started his independent career at FSU in 2002 and moved to WVU in 2016. The current mission of his research program is to impact the drug discovery process by contributing fundamental new knowledge in organic synthesis. The fragmentation / olefination methodology featured herein has figured prominently in this effort over the past few years. |

|
Hyung Joo Kim was born and raised in Daejeon, South Korea. He received his B.S. and M.S. in chemistry from Korea University under the direction of Professor Cheol-Hong Cheon. He is currently pursuing a Ph.D. at the University of Wisconsin-Madison under the supervision of Professor Tehshik P. Yoon. His research focuses on enantioselective ketonization utilizing two different carboxylic acid derivatives via metallaphotoredox reaction. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved