Org. Synth. 2024, 101, 164-180
DOI: 10.15227/orgsyn.101.0164
Difluoromethylation of Phenols
Submitted by Allison T. Hands, Zachary G. Walters, Jacob P. Sorrentino, and Neil K. Garg*
1Checked by Yi Xiao, Milos Vavrik and Nuno Maulide
1. Procedure (Note 1)
A. 1-(3-Chloro-4-(difluoromethoxy)phenyl)ethan-1-one (3). A single-necked (24/40 joint) 100 mL round-bottomed flask is equipped with a magnetic stir bar (2.5 x 1.5 cm, football shaped). The flask is then charged sequentially with 1-(3-chloro-4-hydroxyphenyl)ethan-1-one (1) (3.00 g, 17.6 mmol, 1.00 equiv) (Note 2) and cesium carbonate (8.60 g, 26.4 mmol, 1.50 equiv) (Note 3) (Figure 1A), after which the flask is sealed with a 24/40 rubber septum. The flask is then connected to a Schlenk line via an 18-gauge x 1.5 in needle, and the headspace of the flask is evacuated under vacuum (<0.1 mmHg) for 1 min and backfilled with nitrogen three times (Note 4). Dry DMF (27 mL) (Note 5) and deionized water (3.2 mL) are added sequentially at 23 ℃ to the flask via syringe (18-gauge x 25.4 cm) (Figure 1B) and stirring is initiated (500 rpm).
Figure 1. (A) Solid reagents in flask (B) Mixture after addition of DMF and H2O (photos provided by authors)
The solution is then degassed with nitrogen for 1 h while stirring at 500 rpm (Note 6) (Figure 2A). After 1 h, the rubber septum is removed and sodium 2-chloro-2,2-difluoroacetate (2) (7.51 g, 49.3 mmol, 2.80 equiv) (Note 7) is added in one portion, under a stream of nitrogen. The flask is then equipped with a flame dried air-condenser (40 cm, 24/40 joint), secured with a green Keck clip (3.8/3.2 cm), and sealed with a rubber septum (24/40). The septum at the top of the air condenser is then connected to a Schlenk line via an 18-gauge x 1.5 in needle and a gas outlet terminating in an oil bubbler via an 18-gauge x 1.5 in needle (Note 8) (Figure 2B).
Figure 2. (A) Reaction after being sparged with nitrogen. (B) Apparatus with air condenser (photos provided by checkers)
Nitrogen is flushed through the system for 5 min. The reaction apparatus is then lowered into an oil bath maintained at 120 ℃ and stirred for 2 h (500 rpm) (Figure 3) . Vigorous bubbling begins upon heating the reaction mixture. The system is maintained under positive pressure of nitrogen for the duration of the reaction.
Figure 3. Reaction immediately after being heated to 120 ℃ (photo provided by authors)
After 2 h the reaction is determined to be complete by TLC analysis (Note 9), and the reaction flask is removed from the oil bath and allowed to cool to 23 ℃ (Figure 4A). The mixture is diluted with deionized water (Figure 4B) (40 mL), stirred for 1 min at 300 rpm, and transferred to a 1 L separatory funnel (24/40 joint).
Figure 4. (A) Reaction mixture after cooling to 23 ℃ (B) Reaction after dilution with water (photos provided by authors)
The flask is then rinsed with an additional 60 mL of water, and the rinse solution is transferred to the separatory funnel. The aqueous layer is extracted with hexanes (5 x 100 mL) (Notes 10 and 11) (Figure 5A), and the combined organic layers are washed with a saturated sodium chloride solution (1 x 50 mL) (Note 12) (Figure 5B) and then with a solution of 10% LiCl in H2O (5 x 50 mL) (Note 13) (Figure 5C) .
Figure 5. (A) First hexanes extraction (B) Saturated NaCl solution wash (C) First 10% LiCl solution wash (photos provided by authors)
The organic phase is dried over anhydrous Na2SO4 (30 g) (Note 14) and filtered through a medium-porosity Büchner funnel (150 mL), eluting with hexanes (25 mL) into a 1000 mL round-bottomed flask (Figure 6A). The solution is then concentrated by rotary evaporation under reduced pressure (30 ℃, 120 mmHg). The product is then dried under high vacuum (<1.0 mmHg) for 1 h to provide aryl difluoromethyl ether product 3 as a yellow oil (3.66 g, 16.6 mmol, 94%) (Notes 15, 16, and 17) (Figure 6B).
Figure 6. (A) Filtration into round bottom flask (B) Pure product (photos provided by authors)
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/about/governance/committees/chemical-safety.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
1-(3-chloro-4-hydroxyphenyl)ethan-1-one,
cesium carbonate,
sodium 2-chloro-2,2-difluoroacetate,
N,N-dimethylformamide,
hexanes,
lithium chloride, and
sodium sulfate. The reaction proceeds with significant evolution of gas and an oil bubbler should be used to ensure proper venting of excess pressure from the system.
2. The authors purchased
1-(3-chloro-4-hydroxyphenyl)ethan-1-one from Ambeed and used it as received. The checkers purchased
1-(3-chloro-4-hydroxyphenyl)ethan-1-one (>98%) from TCI and used it as received.
3. The authors and checkers purchased
cesium carbonate (>99%) from Strem and used it as received.
4. While the authors used
nitrogen gas, the checkers used argon gas throughout the procedure.
5. The authors purchased
DMF (>99.5%) from Fisher Scientific. Before use, it was passed through an activated alumina column under argon using a GlassContour solvent purification system. The checkers purchased
DMF (99.8%, Extra Dry over Molecular Sieve, AcroSeal) from Fisher Scientific and used it as received. Upon addition of
DMF and water to
cesium carbonate, an exotherm was observed, reaching approximately 40-45 ℃. No action was taken to prevent this and the reaction mixture returned to ambient temperature within the hour of sparging with inert gas.
6. To degas the solution with
nitrogen, the septum is equipped with a gas outlet 18-gauge x 1.5 cm needle and an 18-gauge x 17 cm
nitrogen inlet needle, which is then submerged beneath the surface of the solution causing
nitrogen to bubble through the reaction mixture.
7. The authors purchased
sodium 2-chloro-2,2-difluoroacetate (>97%) from Ambeed and used it as received. The checkers purchased
sodium 2-chloro-2,2-difluoroacetate (>99%) from TCI and used it as received.
8. The oil bubbler was monitored during this time to confirm the gas outlet was appropriately configured. The reaction proceeds with significant evolution of gas, and an oil bubbler should be used to ensure proper venting of excess pressure from the system.
9. The progress of the reaction is monitored by TLC analysis on silica gel with 1:2
ethyl acetate:
hexanes used as an eluent. To obtain a TLC sample, the air condenser is briefly removed under positive
nitrogen pressure and a drop of reaction mixture is removed and added into a 1 mL vial containing 0.2 mL of
EtOAc and 0.2 mL of 1 M aqueous HCl. The biphasic mixture is shaken and the phases are allowed to separate. The organic phase on top is spotted on the TLC plate. The plate is visualized using a UV lamp (254 nm). The
1-(3-chloro-4-hydroxyphenyl)ethan-1-one (starting material) has R
f 0.3 and the
1-(3-chloro-4 (difluoromethoxy)phenyl)ethan-1-one (
3) has R
f 0.5 (Figure 7).
Figure 7. TLC of the crude reaction mixture after 2 h (SM = 1-(3-chloro-4-hydroxyphenyl) ethan-1-one starting material, Co = co-spot of S and R, and R = reaction mixture) (photo provided by checkers)
10. The authors purchased
hexanes (>98.5%) from Sigma-Aldrich and used it as received. In all cases, instead of
hexanes, the authors used
heptanes which was purchased from Donauchemie and used after distillation.
11. An emulsion forms at the interface of the organic and aqueous layer. The emulsion was collected with the organic layer during the extractions and washes, but drained into the aqueous layer on the final 10%
LiCl solution wash.
12. The authors purchased
NaCl (>99.5%) from VWR and used it as received.
13. The authors purchased
LiCl (>99.5%) from Fischer Scientific and used it as received. The checkers purchased
LiCl (>98%, anhydrous) from Fisher Scientific and used it as received.
14. The authors and checkers purchased anhydrous
sodium sulfate (99.5%) from VWR and used it as received.
15. The product can be characterized as follows:
1H NMR
pdf (700 MHz, CDCl
3) δ: 8.05 (d,
J = 2.1 Hz, 1H), 7.86 (dd,
J = 8.5, 2.1 Hz, 1H), 7.31 (dd,
J = 8.5, 0.9 Hz, 1H), 6.62 (t,
J = 72.5 Hz, 1H), 2.59 (s, 3H);
13C{
1H} NMR
pdf (176 MHz, CDCl
3) δ: 195.6, 150.5 (t,
J = 2.7 Hz), 135.2, 131.2, 128.3, 126.7, 120.7, 115.4 (t,
J = 264.3 Hz), 26.7;
19F{
1H} NMR
pdf (659 MHz, CDCl
3) δ: -82.00 (s, 2F); IR (neat): 1688, 1595, 1494, 1385, 1358, 1251, 1100, 1045, 784; HRMS-ESI
- (
m/z) [M-H]
- calcd for C
9H
6O
235ClF
2: 219.0030. Found: 219.0029; R
f 0.5 (1:2;
ethyl acetate:
hexanes).
16. The purity of
3 was determined to be >97 wt% by qNMR
pdf using
1,3,5-trimethoxybenzene (Sigma-Aldrich, >99%) as the internal standard.
17. The checkers' second reaction, performed on half scale (8.8 mmol), provided 1.78 g (92%) of the product, for which the purity was also determined to be >97 wt% by qNMR using
1,3,5-trimethoxybenzene (Sigma-Aldrich, >99%) as the internal standard.
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Fluorine has distinctive physiochemical properties that have long fascinated chemists. This small and strongly electronegative element can have profound effects on the both the structural and electronic nature of molecules.
2 The implementation of C-F bonds in medicinal compounds has become increasingly common, as they are used to confer desirable pharmacokinetic properties to a bioactive or potential drug molecule.
3,4 One particularly useful fluorine motif, the difluoromethyl ether, has been shown to modulate metabolic stability, lipophilicity, and conformation in several drug development campaigns.
5 Difluoromethyl ethers can also behave as bioisosteres for a number of functional groups.
6 The potential applications of difluoromethyl ethers necessitate robust synthetic strategies for their installation.
The procedure herein offers quick and facile synthetic access to aryl difluoromethyl ethers from phenols through a transformation thought to proceed via a difluorocarbene intermediate. Difluorocarbenes exhibit unique reactivity deriving from their unusual stability and electrophilicity.
7 When fluorine atoms are attached directly to the carbene center they stabilize the carbene in two ways: 1) inductively withdrawing on the carbene's filled σ orbital, and 2) back bonding of fluorine's lone pair into the carbene's empty p orbital.
8,9 As a result of these "push and pull" forces, difluorocarbene exist as singlet carbenes and are generally more stable than other carbenes and preferentially react with most electron rich nucleophiles. These distinct electronic characteristics have spurred the development of methodologies that harness the useful reactivity of difluorocarbenes.
There are several synthetic strategies for generating difluorocarbene, including nucleophilic activation, organozinc, or gaseous reagents.
10,11 However, these methods often require necessitate harsh conditions or in the case of perfluorocarbons, are toxic and environmentally harmful. By contrast, decarboxylative methods have emerged as an operationally simple alternative. Two major classes of decarboxylative reagents exist: phosphonium salts and halo difluoroacetate salts. While they possess similar reactivity, phosphonium salts are both hydroscopic and sensitive to water whereas halodifluoroacetate salts are commercially available in bulk and indefinitely stable under ambient conditions. They also possess mild toxicity and environmental impact making them an attractive difluoromethylating reagent.
The procedure described herein utilizes sodium chlorodifluoroacetate (
2) as a difluorocarbene precursor. An overview of the presumed mechanism is shown in Figure 8A. Thermal decarboxylation of sodium chlorodifluoroacetate (
2) (generates difluorocarbene (
4). The electrophilic difluorocarbene is then trapped by the phenolate nucleophile, generated under basic conditions from
5, followed by subsequent protonation to access the desired aryl difluoromethyl ether,
3. Notably, the chromatography-free protocol offers quick and facile access to aryl difluoromethyl ethers. In addition to difluoromethyl ethers, this method also provides efficient access to
N-difluoromethyl (
5),
S-difluoromethyl (
6), and
Se-difluoromethyl (
7) groups (Figure 8B).
12Figure 8. (A) Decarboxylative Difluoromethylation; (B) Difluoromethylation of heteroatoms (N, S, and Se)
Furthermore, the general transformation we feature has been frequently utilized by medicinal chemists in drug discovery campaigns. Examples of bioactive molecules (
8-13) containing aryl difluoromethyl ethers accessed using
2 are shown in Figure 9.
13,14,15,16,17 Moreover, the development of a second-generation process route to Roflumilast (
8), a multibillion-dollar drug which treats chronic obstructive pulmonary disease (COPD), highlights the advantages of this transformation versus other difluoromethylation methods.
18 Previously, the group at AstraZeneca accessed aryl difluoromethyl ether
8 using chlorodifluoromethane but found it challenging to apply the reagent due to: (1) formation of dimer and trimer byproducts (2) toxicity and environmental impact of chlorodifluoromethane, and (3) variable yields when using a gas in large scale production. Ultimately, they found sodium chlorodifluoroacetate preferable due to its stability, availability in bulk, and relatively low environmental impact.
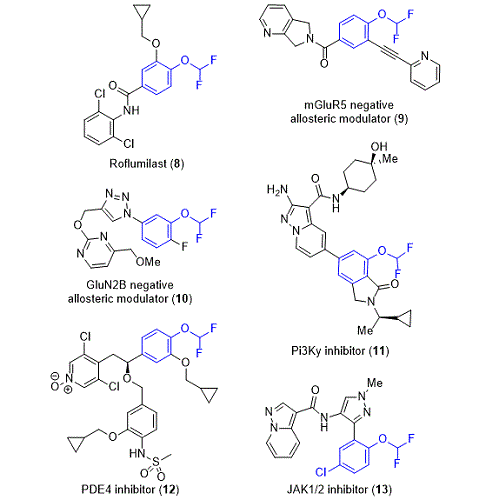
Figure 9. Aryl difluoromethyl ether-containing bioactive molecules 8-13
Finally, in one unique instance the difluoromethyl group was creatively implemented as a protecting group in total synthesis. Recently, the Koert group converted intermediate
16 (Figure 10) to aryl difluoromethyl ether
17. This prevented overoxidation of the chromone ring under harsh oxidation conditions employed later
en route to preussochromomone D (
18).
19 The authors were then able to remove the difluoromethyl group under Lewis acidic conditions and elaborate
17 to the natural product.
20Figure 10. Aryl difluoromethyl ether intermediate in preussochromone D synthesis
This procedure highlights a simple approach to access aryl difluoromethyl ethers from phenols, enabled by the readily available, bench stable, and relatively non-toxic difluorocarbene source, sodium chlorodifluoroacetate. Analogously, the sodium chlorodifluoroacetate reagent can be used to achieve the difluoromethylation of other heteroatoms, including nitrogen, sulfur, and selenium. We hope this procedure will help enable chemists to introduce the important difluoromethyl functional group for a variety of synthetic and medicinal applications.
Appendix
Chemical Abstracts Nomenclature (Registry Number)
1-(3-Chloro-4-hydroxyphenyl)ethan-1-one; (1) (2892-29-7)
Cesium carbonate; (534-17-8)
Sodium 2-chloro-2,2-difluoroacetate;(2) (1895-39-2)
Lithium chloride; (7447-41-8)
Sodium sulfate; (7757-82-6)

|
Allison Hands was born in Los Angeles, CA and raised in Barrington, RI. In 2019, she received her B.A. in Chemistry from Skidmore College where she carried out research under the direction of Professor Steven Frey. In 2022, she then moved to the University of California, Los Angeles, where she is currently a second-year graduate student in Professor Neil K. Garg's laboratory. Her studies primarily focus on developing synthetic methods utilizing strained intermediates. |

|
Zach G. Walters was born and raised in Battle Ground, Washington. In 2022, he received his B.S. in Chemistry from Boise State University, where he carried out research under the direction of Professor Don L. Warner. In 2022, he began his graduate studies at the University of California, Los Angeles, where he is currently a second-year graduate student in Professor Neil K. Garg's laboratory. His studies focus on both total synthesis and methodology. |

|
Jacob P. Sorrentino earned his B.S. in Biology with a minor in Chemistry at Nevada State College, Henderson, NV, USA in 2016, studying under Professor Zachary Woydziak. Jacob then went on to receive his Ph.D. in medicinal chemistry in 2022 from Department of Medicinal Chemistry at the University of Kansas, Lawrence, KS, USA working under Professor Ryan Altman. Jacob is currently working as a postdoctoral researcher in the laboratory of Professor Neil Garg in the Department of Chemistry and Biochemistry at the University of California, Los Angeles, CA, USA. |

|
Neil Garg is the Distinguished Kenneth N. Trueblood Professor of Chemistry at the University of California, Los Angeles. His laboratory develops novel synthetic strategies and methodologies to enable the total synthesis of complex bioactive molecules. |

|
Yi Xiao was born in Zhejiang, China. In 2021, she received her MChem in Chemistry from University of Oxford, where she carried out research under the supervision of Prof. Angela Russell. She then moved to the University of Vienna for her doctoral studies under the supervision of Prof. Nuno Maulide. Her research focuses on the synthesis of novel fluorescent probes and their biological applications. |

|
Miloš Vavrík was born in Trenčín, Slovakia. In 2023, he received his MSc in Chemistry from University of Vienna, where he worked under the supervision of Prof. Nuno Maulide. He then decided to pursue his doctoral studies in the same research group, where he is currently a second-year Ph.D. student. His research focuses on highly reactive electrophilic intermediates and their applications in organic synthesis |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved