Org. Synth. 2024, 101, 366-381
DOI: 10.15227/orgsyn.101.0366
Synthesis of 3,3-Dimethyl-nitroso-2,3-dihydrobenzo[d]isothiazole 1,1-dioxide (NO-1)
Submitted by Keerthana Chakkanalil, Pravien S. Rajaram, and Ryan D. Baxter*
1Checked by Haoran Xiong, Hirofumi Ueda, and Hidetoshi Tokuyama
1. Procedure (Note 1)
3-Chlorobenzo[d]isothiazole 1,1-dioxide (2). A 500 mL two-necked round-bottomed flask is equipped with a thermometer to check the internal temperature and a 4 cm Teflon-coated magnetic stir bar (Note 2). Saccharin (10.0 g, 54.6 mmol, 1.0 equiv) (Note 3) and 1,4-dioxane (250 mL) (Note 4) are added and stirred until the saccharin dissolves. Thionyl chloride (10.0 mL, 138 mmol, 2.5 equiv) (Note 5) is added dropwise over 5 min using a syringe, followed by addition of catalytic dimethylformamide (DMF) (1 mL) (Note 6) after 5 min of stirring. The round-bottomed flask is placed on a heating mantle (Note 7) with sand. The neck is fitted with a 29/32 reflux condenser (Figure 1A, B). The mixture is initially colorless (Figure 1B) and slowly turns yellow (Figure 1C). The reaction reaches a reflux temperature of 101 ℃ in 2 h and is then refluxed for 48 h. After the reaction is complete (Note 8), it is allowed to cool to room temperature (20 ℃) and a clear yellow solution is concentrated under vacuum (18 mmHg) using a rotary evaporator in a water bath at 50 ℃ to remove 1,4-dioxane and excess SOCl2 (Note 9). The white solid, 3-chlorobenzo[d]isothiazole 1,1-dioxide (2) (11.4 g), is used in the next step without further purification (Notes 10 and 11).

Figure 1. A) Reaction set-up fitted with the condenser and placed on the heating mantle with sand; B) Reaction mixture after adding thionyl chloride and DMF; C) Reaction mixture on reaching the temperature 101 ℃. (photos provide by checker)
B. 3,3-Dimethyl-2,3-dihydrobenzo[d]isothiazole 1,1-dioxide (3). A 500 mL three-necked round-bottomed flask is equipped with a thermometer to monitor the internal temperature and an addition funnel capped with a septum. A 4 cm Teflon-coated magnetic stir bar and 2 (8.00 g, 38.3 mmol, 1.0 equiv) are added to the flask (Figure 2A). The third neck is connected to a Schlenk line, and the apparatus is purged with the argon to create an inert atmosphere. Dry diethyl ether (100 mL) (6) is added via 200 mL pressure-equalizing addition funnel (29/32 joint) to the three-necked round-bottomed flask. The reaction mixture is cooled to -10 ℃ in a dry ice/methanol bath (MeOH/H2O:10/90), after which the reaction is then stirred for 10 min. Methyllithium (MeLi) (1.06 M, 150 mL, 159 mmol, 4.0 equiv) (Note 17) is transferred to the addition funnel using a syringe and then added dropwise from the addition funnel with stirring over a time period of 30 min while ensuring the temperature remains below -5 ℃. The reaction mixture is stirred for 1 h at -10 ℃, then the cooling bath is removed and is allowed to warm to room temperature (20 ℃) before stirring an additional 12 h (Note 18).

Figure 2. A) Reaction setup; B) Reaction mixture after adding MeLi; C) sReaction mixture after 2 hours. D) Recrystallized pale yellow crystals, DMBS (3) (photos provide by checker)
The crude reaction mixture is then carefully poured into a 500 mL Erlenmeyer flask containing 1.4 M HCl solution (150 mL), pre-cooled to 0 ℃ with stirring and the resulting mixture extracted with CH2Cl2 (50 mL × 3) using a 1000 mL separatory funnel. The combined organic layer is washed with a saturated NaCl solution (50 mL × 3), dried over Na2SO4 (15 g), and filtered through a funnel with a cotton plug into a 1000 mL round-bottomed flask. The organic layer is concentrated at 25 ℃ under reduced pressure (18 mmHg). After concentration the crude mixture is added to the top of a chromatography column (7.5 cm ID X 42 cm E.L., the height of silica gel is 19 cm) prepared with 300 g of silica gel (Note 13). The column is eluted with 4000 mL of EtOAc/CH2Cl2 (1:20, v/v) (Rf = 0.3) as the mobile phase. The fractions are collected in 25 x 200 mm test tubes (50 mL), and the desired product is obtained in fractions 25-43 (Figure 3). The fractions are concentrated at room temperature (23 ℃) under reduced pressure (18 mmHg). The isolated product, 3,3-dimethyl-2,3-dihydrobenzo[d]isothiazole 1,1-dioxide (DMBS), 3 (3.58 g, before recrystallization) is a light brown solid with minor, colored impurities.
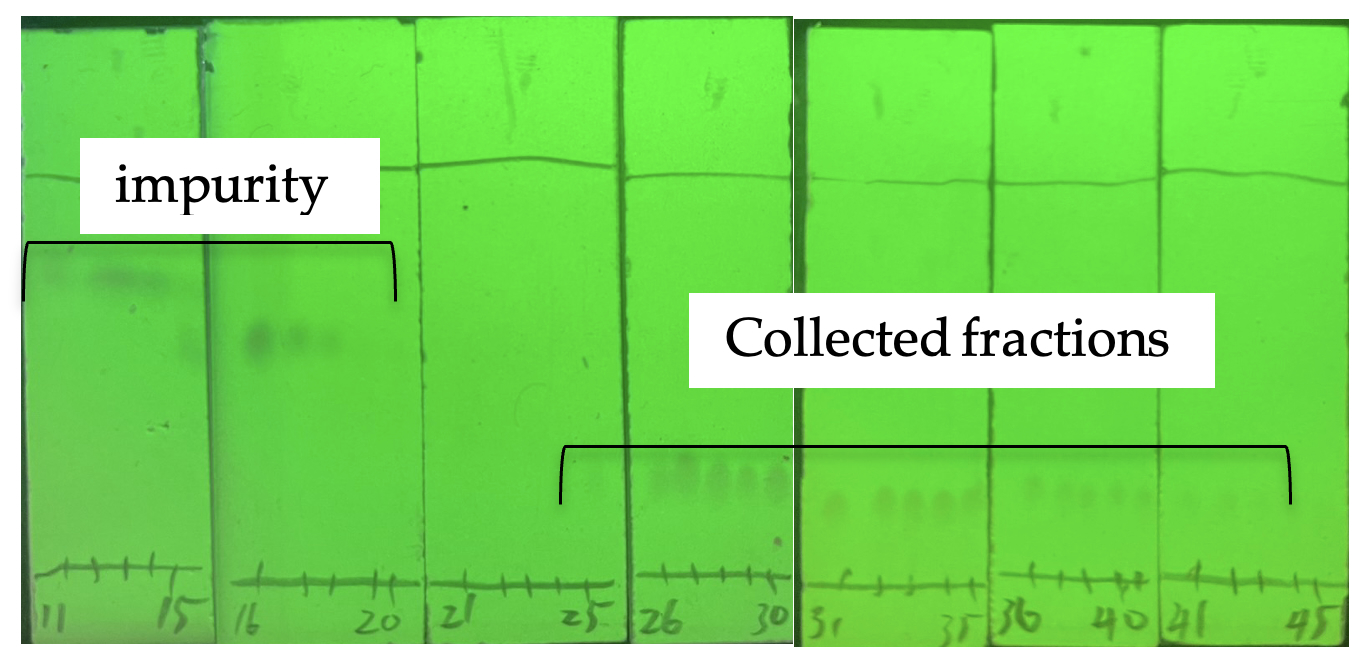
Figure 3. TLC analysis of column chromatography visualized by 254 nm UV (photos provide by checker)
Colored impurities present in compound 3 are removed by recrystallization. The impure compound is dissolved in 10 mL of hot CH2Cl2 (39 ℃), and cold hexane (5 mL) is added in one portion to precipitate the pure compound. The precipitate is filtered with suction through a Büchner funnel and washed with cold hexane to give pale yellow crystals of DMBS (3) (2.94-3.44 g, 39-45% for two steps) (Notes 19 and 20) (Figure 2D).
C. 3,3-Dimethyl-nitroso-2,3-dihydrobenzo[d]isothiazole 1,1-dioxide (4). A 300 mL two-necked round-bottomed flask is used. One neck is equipped with a thermometer to monitor the internal temperature, and the other neck is equipped with 29/42 rubber septum, which is pierced with an 18G vent needle (1.2×38 mm) and left open to the air. To the round-bottomed flask, equipped with a 4 cm Teflon-coated magnetic stir bar, are added DMBS (3) (2.50 g, 12.7 mmol, 1.0 equiv) and p-toluenesulfonic acid monohydrate (pTsOH.H2O) (3.38 g, 17.7 mmol, 1.4 equiv) (Note 21), followed by the addition of 125 mL of CH2Cl2 (Note 22) (Figure 4A). The resulting mixture is stirred at room temperature for 10 min, cooled to 2 ℃ in ice bath (~30 min to reach 2 ℃), and then stirred for an additional 10 min. Sodium nitrite (1.26 g, 18.2 mmol, 1.44 equiv) (Note 23) is added in five equal portions to the cooled mixture over the course of 5 min. The color of the reaction mixture (Figure 4B) changes from light yellow to yellow (Figure 4C) over approximately 30 min. The reaction is warmed to room temperature by removing the cooling bath and is then stirred for 6 h, at which time TLC reveals the reaction is complete (Note 24).

Figure 4. A) Reaction set up; B) Reaction mixture after adding NaNO2; C) Reaction mixture after 30 min; D) NO-1 (4) isolated from the column. (photos provided by checker)
The reaction mixture is filtered through a medium porosity sintered glass funnel (5.5 cm OD × 7.0 cm height) with suction to remove insoluble materials and washed using CH2Cl2 (10 mL). The filtrate is then concentrated at room temperature (23 ℃) under reduced pressure (18 mmHg) and purified with column chromatography using CH2Cl2/hexane (2:1, v/v) (Rf = 0.4) as the mobile phase. After concentration the crude mixture is loaded onto the top of a chromatography column (4.5 cm ID X 42 cm E.L.) prepared with 75 g of silica gel (Note 13). The column is eluted with 750 mL of CH2Cl2/hexane (2:1, v/v) as the mobile phase. The fractions are collected in 15 x 150 mm test tubes (20 mL), and the desired product (Rf = 0.4) is obtained in fractions 10-24 (Figure 5). The isolated fractions are concentrated at room temperature (23 ℃) under reduced pressure (18 mmHg) to obtain a yellow solid product 3,3-dimethyl-nitroso-2,3-dihydrobenzo[d]isothiazole 1,1-dioxide, NO-1 (4) (2.29-2.39 g, 77-81% yield) (Notes 25, 26, 27, and 28) (Figure 4D).

Figure 5. TLC analysis of column chromatography visualized by 254 nm UV (photos provide by checker)
NO-1 (4) is observed as two rotational isomers, NO-1a and NO-1b, that differ in the orientation of the nitroso functional group.
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/about/governance/committees/chemical-safety.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
saccharin,
thionyl chloride,
N,N-dimethylformamide,
1,4-dioxane,
methyllithium,
diethyl ether,
sodium nitrite,
p-toluenesulfonic acid monohydrate,
dichloromethane,
ethyl acetate, hexanes,
methanol, silica, and
1-bromo-3,5-dimethoxybenzene. Caution:
thionyl chloride fumes in moist air and react vigorously with water and
methyllithium catches fire on exposing to air. Caution should be taken when quenching reactions containing
methyllithium, as vigorous reaction with aqueous solutions can take place if not cooled to 0 °C. Nitrososulfonamide,
NO-1 is potentially toxic. The decomposition of the material releases
nitric oxide, which can be harmful if inhaled. Manipulations of the material should be performed in a ventilated fume hood.
2. All reactions were conducted in oven-dried glassware.
3.
Saccharin (
benzo[d]isothiazol-3(2H)-one 1,1-dioxide) (>98%) was obtained from Sigma-Aldrich and used it as received.
4.
1,4-Dioxane, Super Dehydrated was obtained from FUJIFILM Wako Pure Chemical Corporation and used as received.
5.
Thionyl chloride (
SOCl2) (>98.0%) was obtained from Tokyo Chemical Industry Co., Ltd. And used it as received.
6.
N,N-Dimethylformamide (
DMF) anhydrous (99.8%) was obtained from Sigma-Aldrich and used it as received.
7. Glas-col fabric heating mantle with a J-Kem controller (model 210) was used.
8. The starting material (SM), co-spot (Co) and completed reaction mixture (RM) were analyzed by TLC using 50%
ethyl acetate/
hexane mixture and 30%
MeOH/
CH2Cl2 mixture as mobile phases. When using 50%
ethyl acetate/
hexane mixture as mobile phase, the product (
2) was observable under UV light with an R
f of 0.75 (Figure 6).
Figure 6. Thin layer chromatography (TLC) analysis of reaction mixture A) 50% ethyl acetate/hexane mixture were used as mobile phase; B) 70% MeOH/CH2Cl2 mixture were used as mobile phase. (photos provide by checker)
9. Removal of solvents was conducted in a ventilated fume hood to avoid exposure to gaseous
HCl and
SO2.
10. The amount of crude material obtained in 3 runs ranged from 10.5-11.4 g. Characterization data of the non-purified
3-chlorobenzo[d]isothiazole 1,1-dioxide (
2):
1H NMR (600 MHz, CDCl
3) δ: 7.94 (d,
J = 7.2 Hz, 1H), 7.88 - 7.80 (m, 3H).
13C NMR (150 MHz, CDCl
3) δ: 166.1, 140.5, 134.9, 134.4, 129.8, 125.1, 122.4. IR (ATR) 3430, 3033, 3017, 3033, 2970, 2926, 1747, 1716, 1648, 1531, 1450, 1341, 1232, 1200 cm
-1. HRMS (EI
+)
m/z: calcd. For C
7H
4ClNO
2S [M]
+ 200.9651; found 200.9652.
11. For purification, 250 mg of crude
2 was absorbed onto 100 mg of silica gel (
Note 12). This material was dry loaded onto the top of a 250 mL chromatography column (2.0 cm ID X 14 cm E.L.) that was prepared with 15 g of silica gel. The column was eluted with 150 mL of 50%
ethyl acetate/
hexane mixture as the mobile phase (R
f = 0.8). The fractions were collected in 15 x 150 mm test tubes (19 mL), and the desired product was obtained in fractions 3-6. The isolated fractions were concentrated at room temperature under reduced pressure (18 mmHg) to obtain
3-chlorobenzo[d]isothiazole 1,1-dioxide (
2) (210 mg) as a pure white solid product (Notes
13,
14, and
15).
12. Silica gel (Silica gel 60 N, 0.040-0.050 mm, spherical and neutral) was purchased from Kanto Chemical Co., Inc. and used as received.
13. Long-term storage of
2 (crude and purified) should be done under argon at 0 to -4 ℃.
14. Characterization data of the purified
3-chlorobenzo[d]isothiazole 1,1-dioxide (
2):
1H NMR
pdf (600 MHz, CDCl
3) δ: 7.94 (d,
J = 7.2 Hz, 1H), 7.88 - 7.80 (m, 3H).
13C NMR
pdf (150 MHz, CDCl
3) δ: 166.1, 140.5, 134.9, 134.4, 129.8, 125.1, 122.4.
15. The purity of product
2 was determined using
1H qNMR
pdf analysis.
1H qNMR
pdf was performed using a mixture of
2 (10 mg) and
1-bromo-3,5-dimethoxybenzene (10 mg) (Thermo Fisher Scientific Co., Inc., 97%, as an internal standard) in
CDCl3. The purity was determined to be 97 wt%.
16.
Diethyl ether (>98%) was purchased from Sigma-Aldrich and dried over activated molecular sieve 4A.
17.
Methyllithium (1.06 M in
diethyl ether) was obtained from Kanto Chemical Co., Inc. and used as received.
18. TLC analysis of the starting material (SM), co-spot (Co) and reaction mixture (RM) was performed with
CH2Cl2 as mobile phase. R
f value of the product equals 0.25 and was observable under the UV light (Figure 7).
Figure 7. TLC analysis of the reaction mixture (photos provide by checker)
19. Characterization data of the purified
3,3-dimethyl-2,3-dihydrobenzo[d]isothiazole 1,1-dioxide (
DMBS) (
3):
1H NMR
pdf (600 MHz, CDCl
3) δ: 7.74 (d,
J = 7.8, 1H), 7.63 (ddd,
J = 7.8, 7.8, 1.2, 1H), 7.52 (ddd,
J = 7.8, 7.8, 1.2, 1H), 7.40 (d,
J = 7.8, 1H), 4.65 (brs, 1H), 1.66 (s, 6H).
13C NMR
pdf (101 MHz, CDCl
3) δ: 146.0, 135.2, 133.4, 129.1, 122.8, 121.2, 60.9, 29.7. IR (ATR) 3412, 3242, 2977, 1679, 1648, 1542, 1378, 1294, 1173, 1152, 1111, 755 cm
-1. HRMS (FAB)
m/z calcd. for C
9H
12NO
2S [M+H]
+ 198.0589; found 198.0597.
20. The purity of product
3 was determined using
1H qNMR
pdf analysis.
1H qNMR
pdf was performed using a mixture of
3 (10.0 mg) and
1-bromo-3,5-dimethoxybenzene (10.0 mg) (Thermo Fisher Scientific Co., Inc., 97%), as an internal standard in CDCl
3. The purity was determined to be 99 wt%.
21.
p-Toluenesulfonic acid monohydrate (
pTsOH.H2O) was purchased from Thermo Fisher Scientific Co., Inc. and used it as received.
22.
Dichloromethane, anhydrous (
CH2Cl2) (>99.8%) was purchased from Sigma-Aldrich and used as received.
23.
Sodium nitrite (
NaNO2) (>97%) was purchased from Thermo Fisher Scientific Co., Inc. and used it as received.
24. TLC analysis of the starting material (SM), Co-spot (Co) and reaction mixture (RM) was performed with
CH2Cl2 as mobile phase. Product (
4) was observable under the UV light and possessed an R
f of 0.7 (Figure 8).
Figure 8. TLC analysis of the reaction mixture (photos provide by checker)
25. Characterization data of the purified
3,3-dimethyl-nitroso-2,3-dihydrobenzo[d]isothiazole 1,1-dioxide,
NO-1 (
4):
1H NMR
pdf (600 MHz, CDCl
3) δ: 7.85 (d,
J = 7.8 Hz, 1H), 7.80 (dd,
J = 7.8, 7.8 Hz, 1H), 7.64 (dd,
J = 7.8, 7.8 Hz, 1H), 7.52 (d,
J = 7.8 Hz, 1H), 2.00 (brs, 6H).
13C NMR
pdf (150 MHz, CDCl
3) δ: 139.3, 135.3, 130.1, 123.3, 122.0, 121.0, 65.3, 29.6. IR (ATR) 3369, 3082, 3033, 2988, 2926, 2875, 1610, 1578, 1483, 1474, 1458, 1450, 1182 cm
-1. HRMS (EI
+)
m/z calcd. for C
9H
10N
2O
3S [M]
+ 226.0413; found 226.0413.
26. The purity of product
4 was determined using
1H qNMR
pdf analysis.
1H qNMR
pdf was performed using a mixture of
4 (10.0 mg) and
1-bromo-3,5-dimethoxybenzene (10.0 mg) (Thermo Fisher Scientific Co., Inc., 97%) as an internal standard in
CDCl3. The purity was determined to be 99 wt%.
27. DSC data of compound 4: An endothermic response is noted at approximately 70 ℃ to indicate an initial melting point. A second endothermic response is also noted at approximately 90 ℃, likely representing separate melting points for the individual rotamers. The onset of a large exotherm is noted at approximately 180 ℃, indicating thermal decomposition of
NO-1 (
4).
28. Long-term storage of
NO-1 (
4) should be performed using an amber-colored container at 0 to -4 ℃.
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Nitric oxide (NO) is a small molecule of high biological importance. It has been implicated in a range of biological processes including vasodilation,
2 immuneregulation,
3 neurotransmission,
4 and the inhibition of platelet aggregation.
5 Because NO is a gaseous molecule with low water solubility, medicinal applications targeting NO pathways predominately involve organic molecules capable of generating NO in situ via direct bond cleavage, enzymatic processes, or both.
6,7 In addition,
N-nitrosoamines are valuable synthetic intermediates but are also found as potentially toxic contaminants throughout our environment that require detection and remediation.
8 The ability to easily access a variety of structurally diverse nitroso compounds is critical to both fully exploiting potential synthetic and medicinal benefits and developing tools to address toxicity concerns that affect public health.
Traditional methods for nitrosation have involved the use of inorganic nitrites, such as NaNO
2, under strongly acidic conditions to generate electrophilic sources of NO.
9 These methods can be effective for the nitrosation of amides, secondary amines, and certain alcohols but lead to rapid diazotization when reacting with primary amines.
10 In addition, NaNO
2 decomposes under basic conditions, and the requirement for nitrosation at low pH limits the scope of substrates that can effectively participate. Because of this observed limitation, recent synthetic efforts have shifted to using the commercially available
tert-butyl nitrite (TBN) as an electrophilic trans-nitrosation reagent.
11 Unlike inorganic nitrites, TBN does not require strong acidic conditions for
trans-nitrosation, although some nucleophiles require excess TBN to minimize reversible trans-nitrosation with tert-butanol.
12 TBN has been effective for nitrosating amides, secondary amines, and certain alcohols but is known to oxidize primary alcohols under atmospheric conditions.
13,14 Because TBN has been known to undergo both homolytic thermolysis and air-mediated oxidation at room temperature, cryogenic storage under an inert atmosphere is required. As detailed, we have developed a new organic reagent that serves as an attractive alternative to TBN for the trans-nitrosation of nucleophiles under mild conditions.
N-Nitrososulfonamide reagent,
NO-1, is a an easily synthesized crystalline material that maintains long-term integrity under ambient storage conditions (Figure 3D).
15 NO-1 undergoes irreversible trans-nitrosation with a variety of nucleophiles, and the sulfonamide byproduct is easily recovered to and can be used to regenerate
NO-1 with high fidelity. Alkyl alcohols, amines, amides, ureas, and thiols are all effectively irreversibly nitrosated by
NO-1 under mild conditions, resulting in a variety of organic nitroso compounds that were previously challenging to access.
15,16
Appendix
Chemical Abstracts Nomenclature (Registry Number)
Saccharin: 1,2-Benzisothiazol-3(2H)-one 1,1-Dioxide; (81-07-2)
SOCl2: Thionyl chloride; (7719-09-7)
Methyllithium; (917-54-4)
p-Toluenesulfonic acid monohydrate; (6192-52-5)
NaNO2: Sodium nitrite; (7632-00-0)
1-Bromo-3,5-dimethoxybenzene; (20469-65-2)

|
Keerthana is a 2nd year Ph.D. student. She holds an integrated master's degree from the National Institute of Science Education and Research (NISER), India. She currently works under Dr. Ryan Baxter, focusing on the synthesis of analogs of cannabidiolic acid and new organic chemistry methodology. |

|
Pravien is a 4th year Ph.D. student. He came to the United States from Sri Lanka in 2016. He obtained his B.S. and M.S. from Fresno State, where he did research under the guidance of Dr. Qiao-Hong Chen. Currently under Dr. Ryan Baxter, Pravien works on the synthesis of cannabidiol analogs for vasodilator drug candidates, focusing on derivative synthesis of natural products and light-mediated reactions. |

|
Ryan D. Baxter received his B.S. in chemistry from the University of Wisconsin, Madison (2005) and an M.Sc. (2007) and Ph.D. (2010) from the University of Michigan. He performed postdoctoral research at The Scripps Research Institute (2011-2014). He is currently an Associate Professor and Chair of the Chemistry & Biochemistry department at the University of California, Merced. His research interests include single-electron transfers, synthetic methods, and luminescent organic materials. |

|
Haoran is a 2nd year Ph.D. student. He was born in Yamaguchi, Japan in 1998. He obtained his B.S. (2021) and M.S. (2023) from the University of Tohoku (Pharmaceutical Sciences), where he did research under the guidance of Prof. Hidetoshi Tokuyama. His research interests include development of novel cyclization reactions and its application to total synthesis of complex natural products. |

|
Hirofumi Ueda received his Ph.D. (2010) from the Tohoku University under the direction of Professor Hidetoshi Tokuyama. After receiving the Ph.D., he started soon his academic carrier as an Assistant Professor in the same group. In 2018, he was promoted to lecturer and in 2023 to his current position of associate professor. He spent 7 months in 2019 at the University of California, Berkeley as a visiting scholar with Prof. Richmond Sarpong. His research interests center on the development of novel synthetic methodology involving oxidation, and applications to the synthesis of complex alkaloids and nitrogen-containing molecules. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved