Org. Synth. 2024, 101, 395-409
DOI: 10.15227/orgsyn.101.0395
Preparation of S-Methyl Methanethiosulfonate from Dimethyl Sulfoxide Initiated by Oxalyl Chloride
Submitted by Xixuan Zhao, Shuai Peng, Hao Wang, Shuai Huang, Baoguo Sun, Hongyu Tian,*
1 and Sen Liang*
1Checked by Roberto Tinelli, Sophie Woolford and Nuno Maulide
1. Procedure (Note 1)
S-Methyl Methanethiosulfonate (2). A three-necked (19/22) 500 mL round-bottomed flask is equipped with a Teflon-coated magnetic stir bar. A rubber septum with a thermometer is fitted into the left neck of the flask, while the right neck is capped with a rubber septum. A 60-mL pressure-equalizing addition funnel is attached to the middle neck of the flask. The upper end of the pressure-equalizing addition funnel is connected to a vacuum gas adapter, which, in turn, is further connected to a mineral oil bubbler through a latex tube (Figure 1A). The flask is charged with DMSO (71.0 mL, 78.0 g, 1.0 mol) (Note 2) and CH3CN (140 mL) under an air atmosphere (Note 3), which is then placed in a dry ice/acetone cold bath until the internal temperature drops below -15 ℃ (this temperature was then maintained by slow and continued addition of portions of dry ice). Into the pressure-equalizing addition funnel, 30 mL of acetonitrile and then oxalyl chloride (17.1 mL, 0.2 mol, 20 mol%) (Note 4) are added successively. oxalyl chloride solution in acetonitrile is added dropwise (one drop every 3-5 seconds) to the reaction mixture via the addition funnel while controlling the rate of addition to maintain the temperature of the reaction mixture below -10 ℃ (Note 5). Gas evolution is observed in the bubbler during the addition process. After approximately 40 minutes, the addition process is complete. The addition funnel is rinsed with 30 mL of acetonitrile, and the rinse solution is added to the reaction flask. The stirring is continued for an additional 10 minutes in the -20 ℃ cold bath. The flask is then removed from the cold bath and warmed to 25 ℃ (Figure 1B) (Note 6). After removing the addition funnel, a reflux condenser is placed onto the middle neck of the flask. The top of the condenser is then linked to a drying tube containing molecular sieves. Methanol (24.3 mL, 0.6 mol, 60 mol%) (Note 7) is added to the reaction mixture using a 50 mL syringe in one portion (Figure 1C). The flask is immersed in an oil bath set at 80 ℃ for heating (Figure 1D). Reflux begins when the internal temperature reaches 66 ℃ (Note 8). During heating, a white solid gradually precipitates out of the reaction mixture (Note 9), and the color of the solution changes from colorless to yellow. After heating at reflux for 6 h, heating was ceased and the reaction mixture was allowed to return to ambient temperature (Figure 1E) (Note 10).

Figure 1. (A) Set-up of glassware for the reaction; (B) Reaction mixture after the addition of oxalyl chloride; (C) Reaction mixture after the addition of methanol; (D) Reaction set-up at beginning of heating; (E) Reaction mixture after refluxing for 6 hours (Photos provided by submitters)
The reaction mixture is cooled to room temperature, filtered by vacuum filtration using a 150 mL glass Buchner funnel and filter paper (MN615, 4-12μm) to remove the solid, and the filter cake is washed with 100 mL of methylene chloride (Figure 2A). The filtrate is then concentrated under reduced pressure on a rotary evaporator (35-50 ℃ bath, 50 mmHg) (Note 11). The residue (Figure 2B) is dissolved in 100 mL of methylene chloride and transferred to a 500 mL beaker. To the beaker is added 50 mL of brine while stirring; the aqueous phase is acidic with a pH of approximately 1 (Note 12). The solution is neutralized with NaOH solution (2.5 mol/L) until neutrality (Note 13) (Figure 2C). The solution in the beaker is transferred to a 500 mL separatory funnel, and the organic phase is separated (Figure 2D). The aqueous phase is extracted with methylene chloride (50 mL × 3), and the combined organic phases are washed with brine (150 mL). The organic phase is dried over anhydrous sodium sulfate (30 g) (Figure 2E), filtered to remove the drying agent (Note 14) (Figure 2F), and the solvent is removed under reduced pressure on a rotary evaporator (35-50 ℃ bath, 50 mmHg). After concentration to approximately 30 mL, the solution is transferred to a 50 mL single-necked round-bottomed flask (Note 15) and further concentrated on a rotary evaporator to remove as much low-boiling solvent as possible (45 ℃ bath, 50 mmHg), yielding the crude product 23.0 g (Figure 2G) (Note 16). The crude product is distilled under reduced pressure (Figure 2H) (Note 17) to yield 16.3 g (12.9% yield) of methyl methanethiosulfonate (Note 18) as a colorless liquid (Figure 2I), bp 72-74/1.4 mbar (Note 19).

Figure 2. (A) Solid filtration in the reaction mixture; (B) Concentrated reaction mixture; (C) Neutralization; (D) Biphasic reaction mixture post-neutralization; (E) Drying of combined organic mixture with anhydrous Na2SO4; (F) Filtration for drying agent removal; (G) Crude product before vacuum distillation; (H) Vacuum distillation; (I) Pure product. (Photos provided by submitters)
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/about/governance/committees/chemical-safety.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
dimethyl sulfoxide,
acetonitrile,
oxalyl chloride,
methanol,
methylene chloride, and
S-methyl methanethiosulfonate.
2. The checkers purchased
dimethyl sulfoxide (99.9%) from TCI, the submitters purchased it from Innochem. Both batches were used as received.
3. The checkers purchased
acetonitrile (≥99.5%) from Fisher Scientific, the submitters purchased it from Innochem. Both batches were used as received.
4. The checkers purchased
oxalyl chloride (98%) from TCI, the submitters purchased it from Mreda and both batches were used as received.
5. During the addition of
oxalyl chloride in
acetonitrile solution, there was a noticeable exothermic reaction, and fog formed in the headspace of the reaction flask. The rate of addition was carefully controlled to avoid excessive temperature increase, keeping it below -10 ℃. When conducting small-scale experiments (
DMSO, 30 mmol),
oxalyl chloride is typically added at 0 ℃. However, when scaling up the experiment, adding
oxalyl chloride at 0 ℃ results in an exothermic reaction that raises the temperature of the reaction mixture, leading to the formation of
chloromethyl methyl sulfide by-product. Keeping the reaction temperature controlled below -10 ℃ can prevent this issue.
6. To quickly bring the temperature of the reaction mixture back to room temperature, the reaction flask is taken out of the low-temperature bath and placed into a 50 ℃ oil bath. After the temperature of the reaction mixture reaches 25℃,
methanol is added. Alternatively, the checkers also noted that warming in a room-temperature water bath leads to the same outcome.
7.
Methanol (≥99.5%) was purchased from Jingchun and used as received. It is also worth noting that the time of adding
methanol to the reaction system can have a very significant impact on the reaction. After the addition of
oxalyl chloride is complete, removing the reaction flask from the low-temperature bath and adding alcohol under low-temperature conditions can quench the reaction. It is essential to add the
methanol only after the reaction mixture has reached room temperature (about 25 ℃) for the reaction to proceed smoothly.
8. During reflux, the internal temperature was maintained around 66 ℃ (the checkers noted a temperature variation between 65 and 70 ℃).
9. After 30 min of reflux initiation, white solid began to appear in the reaction mixture, and as the reaction progressed, the amount of white solid gradually increased. The color of the reaction mixture changed from initially colorless to yellow over time.
10. The reaction process was monitored by
1H NMR or GC-MS. After refluxing for 6 hours, a small amount of residual
DMSO was observed by
1H NMR or GC-MS. Extending the reaction time does not significantly affect the amount of
DMSO remaining, indicating that the reaction can be terminated after 6 hours. The
1H NMR spectrum of the reaction mixture after refluxing for 6 hours is attached at the end of the article.
11. Due to the presence of malodorous volatile components, it is recommended to perform this concentration step using a rotavap placed in a well-ventilated fume hood.
12. During this reaction,
HCl is released, making the reaction system acidic.
13. Neutralizing to approximately neutral would require about 55-60 mL of
NaOH solution (2.5 mol/L).
14. The drying agent was rinsed with 50 mL of
methylene chloride.
15. In each transfer of the product, the container should be rinsed with solvent to ensure complete transfer of the product.
16. The submitters obtained a crude amount of 20.5 g. A second run carried out on half scale by the checkers yielded 12.1 g of crude product. The crude product was quantified before vacuum distillation using
CH2Br2 (Adamas, 99.0%, used as received) as an internal standard with qNMR
pdf, resulting in a yield of 16.0% in full agreement with the submitters.
17. During the vacuum distillation process, the pressure of the oil pump was maintained about 1.4 mbar, the oil bath temperature was set to 110 ℃, and the fraction temperature was between 72-74 ℃. The first 3 drops of the distillate were discarded, and collection was resumed from the fourth drop. When the temperature began to drop below 72 ℃, collection was ceased.
18. A second run performed on half scale by the checkers gave 8.35g (13.3%). The submitters reported a yield of 17.6 g (14.0%). After vacuum distillation, there was a slight decrease in yield due to a small amount of product remaining in the distillation flask. The purity of product
2 obtained from distillation was determined to be 99.6% by qNMR using 26.0 mg of
2 and 14.6 mg of
dimethyl fumarate (TCI, 98.0%; the submitters purchased from Tan-Mo Technology Co., Ltd, 99.9%, used as received) as the internal standard. The submitters report a purity of 98.9%, as obtained using the same method. The product exhibited the following spectroscopic data:
1H NMR
pdf (600 MHz, CDCl
3) δ 3.31 (s, 3 H), 2.70 (s, 3 H);
13C NMR
pdf (151 MHz, CDCl
3) δ 49.0, 18.5.
19. The observed boiling points were consistent for both checkers. The submitters, however, repeatedly observed a different boiling point which is reported here for additional reference: 82-84 ℃/0.49 mbar.
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
S-Methyl methanethiosulfonate (MMTS) is a highly useful electrophilic methanesulfenylation reagent. In addition to MMTS, commonly used methanesulfenylation reagents also include methanesulfenyl chloride and dimethyl disulfide. Among these three reagents, methanesulfenyl chloride is too reactive, making its preparation and usage inconvenient, while dimethyl disulfide has limited reactivity, resulting in a narrow range of applications. In contrast, MMTS possesses both sufficient reactivity and stability, making it the most commonly used methanesulfenylation reagent. Literature-reported methods for MMTS preparation include the hydrolysis of methanesulfenyl chloride obtained by the oxidation of dimethyl disulfide with chlorine,
2 silver-assisted displacement of disulfide with sodium methanesulfinate,
3 reduction of sulfonyl halides with potassium iodide
4 or activated zinc powder,
5 and oxidation of dimethyl disulfide with microwave-supported permanganate or hydrogen peroxide in AcOH.
6 Additionally, Laszlo and Mathy accidentally discovered that DMSO can be converted into MMTS by treatment with chlorotrimethylsilane and ethylene glycol.
7 Among these methods, the preparation of MMTS by the reduction of methanesulfonyl chloride with Zn powder, as reported by Chemla and Karoyan, is the most reliable and practical method.
5d However, it is evident that the practicality of these methods is still lacking, resulting in MMTS remaining expensive among commonly used reagents.
In our recent study on the application of DMSO/(COCl)
2 combination reagents, we unexpectedly discovered that DMSO undergoes decomposition in the presence of catalytic amounts of (COCl)
2 to produce MMTS (Scheme 1). Methanesulfenic acid is a crucial intermediate in the formation of MMTS. Possible pathways for the formation of MMTS from methanesulfenic acid are discussed in detail in our paper published in
J.
Sulfur Chem. in 2021.
8 Calculated based on the amount of MMTS produced per molecule of DMSO, the yield is around 16%. Compared to the method reported by Chemla and Karoyan,
5d our method has advantages in terms of cost and ease of operation, making it a more practical approach.
Scheme 1. Formation of MMTS from DMSO initiated by (COCl)2
In this work, we made some improvements based on our previously reported method. As illustrated in Scheme 1, the formation mechanism of MMTS involves the release of formaldehyde during the reaction, thus resulting in the observation of a large amount of polymeric formaldehyde as a white solid on the condenser. This makes cleaning of the instrument inconvenient. To prevent the escape of formaldehyde from the reaction system, we attempted to trap it by adding alcohols to the reaction mixture. Initially, we tried ethylene glycol, which captured formaldehyde and formed the corresponding acetal 1,3-dioxolane in the reaction system. Compared to the conditions without alcohol, we observed three changes: (1) the reaction slowed down. For the same amount of DMSO (1 mol), the reaction was completed in approximately 3.5 hours without alcohol, while with the addition of alcohol, the reaction time extended to 8 hours. (2) During the reaction, a white solid precipitated from the reaction mixture. (3) After 8 hours of reaction, a small amount of DMSO remained, and even with prolonged reaction time, it could not be completely consumed. To improve the reaction efficiency, we increased the amount of oxalyl chloride from 10 mol% in the original method to 20 mol%, which allowed consumption of most of DMSO within 6 hours. The white solid in the reaction mixture was identified as NH4Cl through 1H NMR analysis. The 1H NMR spectrum of NH4Cl is attached at the end of the article. The formation mechanism is speculated as follows: after adding alcohol, water molecules are released during the reaction between alcohol and formaldehyde to form acetal, leading to hydrolysis of the solvent acetonitrile to produce acetic acid and ammonia (Scheme 2). Ammonia combines with HCl in the system to form ammonium chloride. Additionally, trace amounts of monoacetyl and diacetyl ester by-products of ethylene glycol were observed in the reaction system, which could not be effectively separated from MMTS during vacuum distillation.
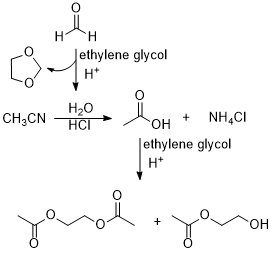
Scheme 2 Formation of NH4Cl and ester by-products derived from the addition of ethylene glycol
We then attempted to use ethanol and methanol to trap formaldehyde. Experimental results showed that methanol was the most effective, without interference from high boiling point by-products, and the product purity was excellent after vacuum distillation.
In summary, this method provides a very convenient route for the preparation of MMTS from DMSO. The reagents used are inexpensive and readily available, requiring no special handling and can be used directly. The low-boiling by-products generated during the reaction process are easily removed, and the product is easily purified, making it advantageous for its simplicity of operation. It is very suitable for large-scale industrial production. This method is expected to contribute to a breakthrough in reducing the market price of MMTS.
Appendix
Chemical Abstracts Nomenclature (Registry Number)
Dimethyl sulfoxide (67-68-5)
Oxalyl chloride (79-37-8)
Methanol (67-56-1)
S-Methyl methanethiosulfonate (2) (2949-92-0)

|
Xixuan Zhao was born and raised in Beijing, China. In 2022, he received his B.S. degree in Applied Chemistry from Beijing Technology and Business University. In 2023, he began his graduate studies at Beijing Technology and Business University, where he is currently a first-year graduate student in Professor Hongyu Tian's laboratory. His studies focus on organic synthetic methodology. |

|
Shuai Peng was born and raised in Beijing. In 2022, he received his B.S. degree in Applied Chemistry from Beijing Technology and Business University. In the autumn of the same year, he began his graduate studies at Beijing Technology and Business University, where he is currently a second-year graduate student in Professor Hongyu Tian's laboratory. His studies focus on the α-oxidation of 1,3-dicarbonyls. |

|
Hao Wang was born and raised in Fuyang City, Anhui Province, China. He earned his B.S. degree in Applied Chemistry from Chizhou University in 2015. He then pursued his graduate studies in Applied Chemistry at Beijing Technology and Business University, beginning in 2016. He completed his Ph.D. degree in 2023 under the supervision of Professor Hongyu Tian. His doctoral research focused on the application of DMSO as a methylene synthon and methylthio synthon in organic synthesis. |

|
Shuai Huang was born and raised in Henan Province, China. He received his B.S. degree in Chemical Engineering and Technology from Zhengzhou University of Light Industry in the summer of 2018, and he completed his M.S. degree under the supervision of Professor Hongyu Tian in 2021 at Beijing Technology and Business University. His research focused on the application of sulfoxide and oxalyl chloride in organic synthesis. |

|
Baoguo Sun is working at Beijing Technology and Business University as a professor of Flavor Chemistry. He is an academician of the Chinese Academy of Engineering. His main research areas include flavor synthesis and flavor analysis. |

|
Hongyu Tian is the Professor of Flavor Chemistry at Beijing Technology and Business University. Her laboratory mainly focuses on the study of synthetic methodology and green synthesis methods for synthetic flavors. |

|
Sen Liang received his PhD from China Agricultural University under the supervision of Professors Chengchu Zeng and Baoguo Sun in 2018. Then he joined Beijing Technology and Business University, and is currently working there as an associate professor of flavor chemistry. His research interests focus on developing electrosynthetic methods for flavor synthesis and creating new types of flavors. |

|
Roberto Tinelli earned his MSc from the University of Pavia in 2020 under the guidance of Prof. Ravelli, specializing in the development of novel substrates for visible light-driven Paternò-Büchi reactions. He has collaborated with the Goodman Group and the Vendruscolo group at the University of Cambridge, gaining proficiency in computational chemistry and programming. Beginning his PhD journey at the University of Vienna in early 2021 under the mentorship of Prof. Maulide, Roberto has worked on the development of new charge-accelerated rearrangements with sulfinamides and various redox-neutral synthetic methods. |

|
Sophie Woolford was born in Oxford, UK and received her MChem (2019) from the University of Manchester under the supervision of Dr Roger Whitehead, during which time she also spent a year at the University of British Columbia carrying out research with Prof. Marco Ciufolini. She then moved to the University of Glasgow where she carried out her PhD (2023) in the group of Prof. Stephen Clark. Currently, she is a postdoctoral fellow at the University of Vienna in Prof. Nuno Maulide's group, where her research focuses on entropy-orientated drug design. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved