Org. Synth. 2024, 101, 488-507
DOI: 10.15227/orgsyn.101.0488
Hydrodealkenylative Cleavage of C(sp3)-C(sp2) Bonds: Preparation of (1S, 3R)-3-Methylcyclohexan-1-ol
Submitted by Brady W. Dehnert and Ohyun Kwon*
1Checked by Matthew Furry, Ryan R. G. Barrett, and Christopher D. Vanderwal
1. Procedure (Note 1)
(1S, 3R)-3-Methylcyclohexan-1-ol (2). A single-necked (24/40 joint) 500 mL round-bottom flask was equipped with a Teflon-coated magnetic stir bar (2.5 x 0.5 cm, pill-shaped). To the flask was added (-)-isopulegol (1) (8.5 mL, 7.7 g, 50 mmol, 1.0 equiv) (Notes 2 and 3) via syringe. Then, MeOH (200 mL, 0.25 M) (Note 4) is added at 23 ℃ to the flask from a graduated cylinder. The mixture was stirred (500 rpm) briefly at room temperature until a homogenous mixture is achieved. The flask was then placed in a saturated dry ice/Acetone bath (400 mL) (Notes 5) (1000 mL dewar) and cooled to -78 ℃ while open to the air. Ozone (Notes 6 and 7) was bubbled through the clear colorless solution (Figure 1A) for 50-100 minutes (Note 8), until complete consumption of the starting material had occurred (Figure 1B). Complete consumption can be indicated by a faint blue color (Note 9). The solution was then sparged with argon (tank pressure 11 psi) through a 16 G needle submerged in the reaction for 20 minutes to expel the excess ozone (Figure 1C).

Figure 1. A. Bubbling of O3/O2 mixture into reaction flask; B. Blue color indicates completion; C. Argon sparge returns clear colorless solution (Photos provided by authors)
Benzenethiol (7.7 mL, 8.3 g, 75 mmol, 1.5 equiv) (Notes 10 and 11) was then added by syringe as a gentle stream over one minute to the clear, colorless solution with stirring at 800 rpm (Figure 2A). This was followed by the portion-wise addition of ferrous sulfate heptahydrate (16.7 g, 60 mmol, 1.2 equiv) (Note 12) (Figure 2B) over 2 minutes by pouring through a powder funnel.
Figure 2. A. Addition of benzenethiol; B. Addition of iron(II) sulfate heptahydrate (Photos provided by authors)
The green heterogeneous mixture was stirred (800 rpm) at -78 ℃ for 15 minutes before the cooling bath was removed (Figure 3A). After the solution warmed to -20 ℃ over the next 10 minutes, the iron sulfate heptahydrate began to dissolve. After warming to room temperature over 2 hours, the heterogeneous orange-brown reaction was stirred for an additional 12 hours (Figure 3B) until complete peroxide reduction, as indicated by TLC (Note 13) and a rusty-brown color. Water (100 mL) (Note 14) was added over 5 minutes at room temperature (Figure 3C) from a 20 mL syringe. A 7 ℃ increase was noted with a thermometer.
Figure 3. A. Reaction after stirring at -78 ℃ for 15 min; B. Reaction after stirring for 12 hours at room temperature; C. After quenching with water (Photos provided by authors)
The heterogeneous orange mixture was transferred to a 1000 mL separatory funnel and the red aqueous layer was extracted with dichloromethane (3 x 150 mL) (Note 15) (Figure 4A). Insoluble emulsions in the first organic extraction were kept with the yellow organic layer, then the combined organic fractions were washed with brine (100 mL) (Note 16), which resolved the emulsions and resulted in an orange brine layer (Figure 4B) (Note 17). The pale yellow organic extract was then dried over anhydrous sodium sulfate (150 g) (Note 18) for 30 min, filtered through a funnel plugged with cotton, and diluted with 500 mL of pentane to facilitate methanol removal in vacuo (Note 19). The organic extract was concentrated in a 2000 mL round-bottomed flask by rotary evaporation under reduced pressure (0 ℃, 500 mmHg to 50 mmHg) to yield 22-32 g of a yellow oil or heterogeneous oily mixture with white solids, which reverted to a yellow oil on standing at room temperature or with gentle warming (Note 20) (Figure 4C and D).
A slurry is prepared from silica (800 g) (Note 21) and a 15% solution of ethyl acetate in hexanes (2000 mL) (Notes 22 and 23) and is loaded into a fritted glass column (inner diameter 8.5 cm x 36 cm long). After packing the column using pressurized air to move the prepared solvent through, the crude material was loaded onto the column with a glass pipette using a minimal amount of hexanes (5 mL). The round-bottom flask was rinsed with hexanes (3 x 5 mL), the rinses were loaded onto the column, the solvent was pushed into the silica using pressurized air, the column wall was rinsed once more with hexanes (5 mL), and sand (400 g) (Note 24) was poured on top (Figure 4E). Solvent left over from packing the silica gel, followed by an additional quantity of 15% ethyl acetate:hexanes (1800 mL), was then flushed through the column into a 4 L jug at a rate of 5 mL/second in order to elute an initial yellow band of diphenyl disulfide.

Figure 4. A. First extraction with dichloromethane; B. Brine wash; C. Pale yellow crude oil; D. Alternative crude obtained as a white solid and pale yellow oil mixture; E. First column loaded with silica, crude, and sand; F. Second column after loading (Photos provided by checkers)
The mobile phase was then increased to 30% ethyl acetate:hexane (4000 mL) and fractions of 65 mL were collected in 2.5 cm x 20 cm test tubes at a rate of 5 mL/second, which afforded 60 fractions. Fractions 42-50 containing pure product were identified by TLC analysis (Notes 25 and 26), collected in a 3000 mL round-bottomed flask, and concentrated by rotary evaporation (0 ℃, 500 mmHg to 50 mmHg). Impure product, identified by TLC in fractions 37-41 and 51-54, was collected in a 1000 mL round-bottomed flask and concentrated by rotary evaporation (0 ℃, 500 mmHg to 5 mmHg).
The concentrated impure material (1.45-1.80 g) was then loaded onto silica gel (230 g) in a second column (inner diameter 5.5 cm x 30 cm long), packed as previously described using 1000 mL of 15% ethyl acetate:hexanes, and sand (70 g) was added (Figure 4F). Solvent left over from packing the silica gel, followed by 30% ethyl acetate:hexanes (1400 mL), were used to elute at a rate of 1.5 mL/s into 1.8 cm x 15 cm test tubes of 25 mL each. Fractions 54-64 containing pure product were identified by TLC analysis (Notes 25 and 26), added to the 3000 mL flask with the pure fractions from the previous column, and concentrated by rotary evaporation (0 ℃, 500 mmHg to 5 mmHg). The resulting oil was transferred to a 5 dram vial using hexanes (3x3 mL) and concentrated by rotary evaporation (0 ℃, 500 mmHg to 5 mmHg) followed by evaporation for 2 hours (0 ℃, 5 mmHg) to yield pure (1S, 3R)-3-methylcyclohexan-1-ol (2) (3.38-3.94 g, 59-69% yield, >97% purity by quantitative NMR) as a clear, colorless oil (Notes 27,28,29,30) (Figure 5).

Figure 5. Isolated (1S, 3R)-3-methylcyclohexan-1-ol (Photo provided by authors)
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/about/governance/committees/chemical-safety.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
(-)-isopulegol,
iron(II) sulfate heptahydrate,
benzenethiol,
methanol,
ozone,
dichloromethane, silica gel,
hexane,
ethyl acetate,
sodium chloride,
sodium sulfate,
1,3,5-trimethoxybenzene, deuterate chloroform, as well as the proper procedures for chromatographic purifications and handling organic peroxides. Organic peroxides are known to be explosive in some cases. It is important that reproducers minimize their risk of danger with this methodology. This includes not over-heating the peroxide, reducing the risk of shock to the reaction mixture, and utilizing a blast shield when doing large scale reactions.
Ozone is an extremely toxic and reactive oxidant that an react with some compounds to form explosive and shock-sensitive products. Although we have not encountered any
ozone-related safety issues in our lab, reaction with
ozone should be performed only by properly trained individuals in a well-ventilated fume hood (use of a blast-shield is also recommended, especially for reactions performed on larger scales).
2.
(-)-Isopulegol (>98%) was purchased from Sigma Aldrich.
3. Commercial
(-)-isopulegol (100 mL) was purified by simple distillation from a 250 mL round-bottomed flask immersed in an oil bath (bath temperature 105 ℃) through a 12 cm Vigreux column (standard taper joint 14/20) prior to use (boiling point 66-67 ℃ at 5.0-7.0 Torr).
4.
Methanol (99.8%) was purchased from Fisher Scientific and was used as received. The checkers purchased the same grade from Sigma Aldrich.
5.
Acetone (99.5%) was purchased from Fisher Scientific and was used as received. The checkers purchased the same grade from Sigma Aldrich.
6. The checkers performed ozonolysis using two different ClearWater Tech Inc. m-1500
ozone generators (1-2 SCFH, 100% power, O
2 feed gas) (Figure 6). The authors performed ozonolysis using a Globalozone GO-d3G (3 g/h)
ozone generator (2.0 L/min, 100% power, O
2 feed gas) (Figure 7). It is worth noting that differences in the ratio of O
3 to O
2 may vary among instruments and will vary ozonolysis times which may affect yields.
Figure 6. One of the ozone generators used by the checkers, with ozone supplied through the top right inlet to the instrument with blue tubing, and white tubing supplying output ozone gas mixture to the reaction vessel. A glass pipette, broken off above the stem for wider aperture, is firmly inserted into a short section of appropriately sized tygon tubing and submerged in the reaction (Photo provided by checkers)
Figure 7. The authors' ozone generator. A. Oxygen source connected to a tygon tubing line; B. ozone generator with tygon tubing on both input and output openings; C. Tygon tubing connected to two deconstructed 1 mL syringe casing held together by parafilm (Photo provided by authors)
7. Alternatively for large scale reactions, the reaction can occur under a closed system where the
ozone is quenched with a 10%
potassium iodide trap. A two-necked (24/40 joints) 500 mL round-bottom flask was equipped with a Teflon-coated magnetic stir bar (2.5 x 0.5 cm, pill-shaped) was used instead where one joint was for bubbling in
ozone and other was for exiting
ozone. The exit port was attached to two sequential 10%
potassium iodide traps. A color change to yellow and dark orange indicates that
ozone is being quenched successfully (Figure 8).
Figure 8. A. Reaction set-up for ozone quenching before ozone bubbling B. Ozone quenching at the completion of the ozonolysis with noticeable color change in traps (Photos provided by authors)
8. The checkers used two different instruments of the same brand, and found one gave consistent reaction times of 50 minutes while the other required 1 hour 40 minutes for completion. The authors reported a reaction time of 1.5 hours.
9. The progress of the ozonolysis was monitored via thin-layer chromatography (TLC) analysis on silica gel with 20%
ethyl acetate/hexanes as the eluent. (Figure 9). The TLC plates were stained with ceric ammonium molydate (CAM). Some substrates are sensitive to being in the presence of
ozone for too long, although this is not the case with
(-)-isopulegol. Monitor the reaction closely with TLC analysis and stop ozonolysis immediately when there is complete consumption of the starting material.
Figure 9. A. TLC of crude ozonolysis reaction after 30 minutes. SM = starting material (SM Rf = 0.46), CS = co-spot of SM and reaction mixture, RXN = reaction mixture (product Rf = 0.17); B. TLC of crude ozonolysis reaction after complete consumption of starting material (Photos provided by authors)
10.
Benzenethiol (99%+) was purchased from Thermo Scientific and was used as received. The checkers purchased
benzenethiol (99%) from Acros.
11. Thiol vapors may contaminate gloves during handling, and gloves used should not be brought outside the fume hood. Also any glassware coming into contact with thiols or disulfides will have stench, and treatment with dilute bleach is recommended for cleaning the reaction vessel, separatory funnel, erlenmeyer flasks used for extraction, column, and any test tube fractions used to collect disulfide.
12.
Iron(II) sulfate heptahydrate (98%) was purchased from Thermo Scientific and was used as received. The checkers purchased
iron(II) sulfate heptahydrate (ACS grade) from Oakwood Products.
13. The progress of the reduction and functionalization was monitored by TLC analysis on silica gel with 20%
ethyl acetate/hexanes as the eluent (Figure 10).
Figure 10. A. TLC of crude reaction for iron(II)-mediated reduction and functionalization. I = intermediate (hydroperoxide Rf = 0.17), CS = co-spot of I and reaction mixture, Rxn = reaction mixture (product Rf = 0.36); B. TLC of SM versus P = purified product in 30% ethyl acetate/hexane (SM Rf = 0.61 and P Rf = 0.42) (Photos provided by authors)
14. In-house deionized water was used as received.
15.
Dichloromethane (99.5%) was purchased from Fisher Scientific and was used as received. The checkers purchased the same grade from Sigma Aldrich.
16. Saturated
sodium chloride solution was made from adding
sodium chloride to in-house deionized water until solvation no longer occurs.
17. The checkers modified the extraction procedure to adequately deal with the significant emulsions initially present on workup. The authors used straightforward extraction and noted in personal communication that they did not shake the separatory funnel, but gently tilted it back and forth then waited several minutes for separation.
18.
Sodium sulfate (99%) was purchased from Fisher Scientific and was used as received.
19.
Pentane (98%) was purchased from Sigma Aldrich and was used as received.
20. The submitted procedure called for concentration at 15 ℃ without the addition of
pentane, but the checkers found that product was identified in the rotary evaporator trap at this temperature and that yield increased with concentration at 0 ℃. Furthermore, the significant
methanol content in the crude sample obtained was found to be highly variable by the checkers. Fortunately, the addition of
pentane was found to assist in removal of the
methanol in vacuo and produced more consistent mixtures for chromatography.
21. Silica gel (40-60 μm, 60 Å, irregular) was purchased from Agela and was used as received. The checkers purchased silica gel F60 (40-63 μm, 60 Å, irregular) from Silicycle.
22. Hexanes (98.5%) was purchased from Fisher Scientific and was used as received. The checkers purchased the same grade from Sigma Aldrich.
23.
Ethyl acetate (99.5%) was purchased from Fisher Scientific and was used as received. The checkers purchased the same grade from Sigma Aldrich.
24. Sand (Sea Washed) was purchased from Fisher Scientific and was used as received.
25. TLC plates (25x65 mm) were fully developed in 30%
EtOAc/Hexanes and visualized with
p-anisaldehyde stain in both separations (Figure 11). Note that there are two spots observed just above the desired product and one spot just after. Fractions containing only the spot just above the desired product were collected with pure material and minimally affected purity. All tubes were rinsed with DCM (3x3 mL) and the rinses concentrated with the fractions.
Figure 11. A. Fractions produced by the first separation. B. TLC analysis of the first separation. C. Fractions produced by the second separation. D. TLC analysis of the second separation (Photos provided by checkers)
26. The authors followed a different TLC protocol during chromatography. TLC plates were run 3x with drying in between each run for better separation of impurities. It is worth noting purity was an issue during the checking process, and it could be attributed to the appearance of a fleeting white impurity when staining with CAM (Figure 12).
Figure 12. A. Fraction tubes that correspond with TLC lanes in Figure 10B. B. TLC of collected fractions from column, here the mobile phase is 15% EtOAc in hexanes ran 3x. C. Appearance of impurity that stains lightly white with CAM staining (Photos provided by checkers)
27.
1H NMR
pdf a pdf b pdf c (500 MHz, CDCl
3) δ: 3.56 (app tt,
J = 10.9, 4.3 Hz, 1H), 1.98-1.90 (m, 2H), 1.77-1.69 (m, 1H), 1.64-1.52 (m, 1H), 1.47 (br s, 1H), 1.47-1.36 (m, 1H), 1.26 (app qt,
J = 13.3, 3.4 Hz, 1H), 1.10 (app qd,
J = 12.1, 3.2 Hz, 1H), 0.91 (d,
J = 6.6 Hz, 3H), 0.87 (app q,
J = 11.8 Hz, 1H), 0.77 (app dq,
J = 12.6, 3.6 Hz, 1H).
13C NMR
pdf (101 MHz, CDCl3) δ: 70.8, 44.7, 35.5, 34.2, 31.5, 24.3, 22.5. IR
pdf (neat liquid) ν
max: 3335 (broad), 2925, 2856, 1450, 1364, 1100, 1045, 1021, 938 cm
-1. bp = 170 ℃ at 760 mmHg. HRMS (EI+) m/z [M]
+ calcd for C
7H
14O 114.1045, found 114.1045. Optical Rotation: [α]
20D -1.4 (
c 1.00, CHCl
3).
28. The authors reported 4.77 g (84%) and 4.83 g (85%) of the product with purities of >98% on identical scale. Yields from the checkers are from three separate runs. Each checker ran the full experiment independently, and two different ozonolysis setups were used in order to show robustness (
Note 7).
29. The purity of
2 was determined by qNMR
pdf a pdf b pdf c in CDCl
3 in all cases with the use of
1,3,5-trimethoxybenzene (
Note 25) as the standard.
30.
1,3,5-trimethoxybenzene (≥99.9%) was purchased from Sigma-Aldrich and was used as received.
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Alkenes are one of the most common functionalities in natural products.
2 With their abundance and utility in natural product synthesis, many transformations exist in the literature that seek to cleave, build, and functionalize olefins. While many reactions seek to cleave the C(sp
2)-C(sp
2) bonds of olefins, the adjacent C(sp
2)-C(sp
3) is seldom cleaved. The Kwon group has recently developed a method to break alkenyl C(sp
2)-C(sp
3) bonds and functionalize the resulting alkyl radical.
3,4 The most notable has been installing C(sp
3)-H bonds,
5,6 but others include C(sp
3)-SR,
7 C(sp
3)-OR,
8 C(sp
2)=O,
8 C(sp
3)-C(sp
2),
9 C(sp
3)-C(sp),
10 and C(sp
3)-NR
2 motifs (Scheme 1).
11Scheme 1. Dealkenylative functionalizations available since 2018.
The forementioned dealkenylative functionalization method involves synthesizing an α-alkoxyhydroperoxide from alkenes by the Criegee ozonolysis (Scheme 2).
12 A (3+2) and retro-(3+2) cycloaddition with ozone will form a carbonyl oxide, known as the Criegee intermediate, that is neutralized by methanol. After formation of the α-methoxyhydroperoxide, addition of an iron(II) salt will reduce the O-O bond to a hydroxyl anion and an alkoxyl radical. The alkoxyl radical will undergo a β-scission event to produce the alkyl radical. In the case of hydrodealkenylation, the alkyl radical will abstract a hydrogen from a hydrogen donor.
Scheme 2. Mechanism for hydrodealkenylation
From organic peroxides, radical hydrogen abstraction has been demonstrated from decades ago. The following reactions were primarily focused on the decomposition of peroxides and analysis of the products. Kharasch demonstrated that methyl radicals excised from cumyl hydroperoxide could abstract hydrogen from dextrose in heptane to produce methane gas in 1950 (Scheme 3).
13 Also in 1950, Hawkins showed that 1-methylcyclopentyl hydroperoxide could be reduced with iron(II) salts to produce 2-hexanone.
14Scheme 3. Peroxide reduction of hydroperoxides and radical hydrogen abstraction
The hydrodealkenylation method was initially used in the synthesis of bicyclic phosphines, CarvoPhos.
5 The isopropenyl moiety in the
endo-face of the catalyst
4' precluded the formation of
endo-CarvoPhos, so the group considered removal of the alkene of dimesylate intermediate
3 at the C-4 position (Scheme 4). They employed ozone in methanol, then iron(II) sulfate heptahydrate and benzenethiol, to successfully cleave the alkene from the dimesylate intermediate
3. The dealkenylated phosphine catalyst could then be produced from the dealkenylated product
5 in both
exo- and
endo-aryl-substituted forms,
6 and
6' respectively, and the
endo-aryl-CarvoPhos
6' would go on to successfully catalyze allene-imine [3+2] annulations.
Scheme 4. Synthesis of CarvoPhos featuring hydrodealkenylation
Later, the same method would be used on a wide variety of readily available or easily synthesized alkenes.
6 One of the most notable examples is the transformation of (-)-isopulegol to (1
S, 3
R)-3-methylcyclohexan-1-ol (Scheme 5). At the time of publication, the abundant alkene (-)-isopulegol could be bought for $14/mol and the dealkenylated product could be bought at $193k/mol. The non-trivial product is a key intermediate to a pharmaceutical agent that deals in androgen modulation.
Scheme 5. Hydrodealkenylation of (-)-isopulegol
The hydrodealkenylation method has been utilized in the total synthesis of three natural products since its publication in 2019. The first to be published in a peer-reviewed journal was by the Liu and Qin in the synthesis of vilmoraconitine.
15 A tricyclic cycloalkene was opened to the resulting aldehyde that would be needed for the later reductive amination step in the synthesis (Scheme 6). The second synthesis was of (+)-shearilicine by the Newhouse group. They reported first to Chemrxiv in 2022, but it was later seen in a peer-reviewed journal in 2023.
16 From a tricyclic core, the isopropenyl functionality that is a remnant of the (-)-carvone starting material was smoothly cleaved. The most recent use of hydrodealkenylation is from the Ito group in 2024 in the asymmetric synthesis of (-)-lemnalemnane A. The alkynylated starting material derived from (
S)-carvone underwent hydrodealkenyaltion to show another example of cleavage of the isopropenyl functionality.
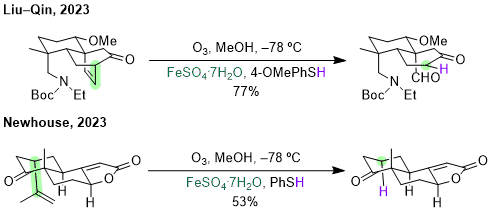
Scheme 6. Uses of hydrodealkenylation in recent natural product syntheses
Hydrodealkenylation and other functionalization strategies are powerful methods to transform readily available alkenes into valuable and useful intermediates. Dealkenylative functionalization is one of the few methods to break the seldom-cleaved C(sp3)-C(sp2) while using readily available ozone to form peroxides and abundant earth metal complexes for the reduction of peroxides. The utility is demonstrated with the simple cleavage of the alkene from (-)-isopulegol to a useful, chiral intermediate.
Appendix
Chemical Abstracts Nomenclature (Registry Number)
(-)-Isopulegol: (1R, 2S, 5R)-5-methyl-2-(prop-1-en-2-yl)cyclohexan-1-ol; (89-79-2) (1)
Benzenethiol; (108-98-5)
Iron(II) Sulfate Heptahydrate; (7782-63-0)

|
Ohyun Kwon, Professor of Chemistry and Biochemistry at UCLA, received her B.S. and M.S. degrees from Seoul National University in 1991 and 1993, respectively. After obtaining her Ph.D. from Columbia University in 1998, and a postdoctoral stint at Harvard University, Kwon began her independent career at UCLA in 2001. Her research involves the development of defunctionalative C-C activation and phosphorus organocatalysis processes and their application in the synthesis of compounds of biological significance. She has played key roles in establishing phosphinocatalysis as one of the main areas of organocatalysis, and is recognized as one of the leaders in the field. |

|
Brady Dehnert was born and raised in Texas. In 2021, he received his B. S. degree in Chemistry from the University of Oklahoma where he worked for Professor Shanteri Singh. In 2021, he began his graduate studies at the University of California, Los Angeles where he is currently a third-year graduate student in Professor Ohyun Kwon's laboratory. |

|
Matthew Furry earned his B.S. in Chemistry from Stockton University in 2022, where he studied the synthesis and photophysics of aza-dipyrrins with Prof. Barry C. Pemberton. Currently, he is a graduate student at the University of California, Irvine where he studies radical approaches to diterpene natural products with Prof. Christopher D. Vanderwal. |

|
Ryan Barrett completed his B.Sc. in Chemistry at the University of British Columbia in 2013, where he conducted research under the supervision of Prof. Harry Brumer III on bio-informatic identification, expression, and synthetic use of galactose oxidase enzymes toward cellulose-adsorbed elastase sensors. He completed his Ph.D. work with Prof. James L. Gleason at McGill University in 2022, where he synthesized hybrid inhibitors for potential treatment in prostate cancer and developed an organocatalytic oxy-Cope / Michael cascade reaction. He is currently a postdoctoral scholar at UC Irvine in Prof. Chris Vanderwal's lab, working in the field of natural product synthesis. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved