Org. Synth. 2024, 101, 524-541
DOI: 10.15227/orgsyn.101.0524
meta-C-H Halogenation of Pyridines, Quinolines, and Isoquinolines: meta-Chlorination of 2-Phenylpyridine
Submitted by Pengwei Xu
1 and Armido Studer*
1Checked by Alexander Sailer and Dirk Trauner
1. Procedure (Note 1)
A. Trimethyl 2-methyl-6-phenyl-2H,9aH-pyrido[2,1-b][1,3]oxazine-2,3,4-tricarboxylate (1). A 250-mL single-neck round-bottom flask, equipped with a Teflon-coated magnetic stir bar (2 cm x 0.5 cm), is charged with 2‐phenylpyridine (3.88 g, 25.0 mmol, 1.0 equiv) (Note 2), and methyl pyruvate (3.06 g, 30.0 mmol, 1.2 equiv) (Note 3), which is dissolved in CH3CN (50 mL) under ambient atmosphere (Note 4). Dimethyl acetylenedicarboxylate (4.26 g, 30.0 mmol, 1.2 equiv.) (Note 5) is added dropwise over three minutes to the stirred reaction mixture using a syringe and the reaction mixture turned slightly yellow immediately (Figure 1A). Then, the reaction vessel is loosely plugged with a glass stopper. After 36 hours of stirring (500 rpm), the reaction mixture turns dark brown (Figure 1B). Afterwards, the reaction flask is connected to a rotary evaporator and all volatiles are removed (50 ℃, 140 mm Hg, then 10 mm Hg). The remaining residue is further dried under high vacuum (<1.0 mmHg, 30 min) (Figure 1C).

Figure 1. (A) Set-up of the reaction after addition of dimethyl acetylenedicarboxylate; (B) Brown solution of the reaction mixture after 36 hours reaction time; (C) Remaining residue after evaporation under high vacuum (<1.0 mmHg) (Photos provided by authors)
The residue is subjected to flash column chromatography. A glass column (4 cm diameter) is wet-packed with silica gel (100 g) suspended in pentane to a total height of 15 cm (Figure 2A, Note 6 and 7). The brown residue is dissolved in CH2Cl2 (15 mL) (Note 8), and loaded onto the silica gel (Figure 2B). The flash column chromatography is run with a slight overpressure of argon. After the dissolved product is fully absorbed onto the silica, the silica gel filling is topped off with sea sand (3 cm) (Note 9). The column is first eluted with 200 mL pentane followed by 300 mL eluent mixture (pentane/EtOAc 5:1) and the eluting solution is discarded (Figure 2 C, Note 10). Afterwards, the column is sequentially charged with 800 mL eluent mixture (pentane/EtOAc 3:1) and 500 mL eluent mixture (pentane/EtOAc 2:1), respectively. The fractions are collected in 100-mL test tubes, filled to 90 mL volume. The fractions 4-12 are combined into a 1 L round-bottom flask and all volatiles are removed by rotary evaporation (50 ℃, 650 mm Hg, then 180 mm Hg) (Note 11). The product is transferred to a 250-mL round-bottom flask with 100 mL CH2Cl2 (Note 8), again concentrated on rotary evaporation (50 ℃, 650 mm Hg, then 10 mm Hg) and dried under high vacuum (<1.0 mmHg) for 1 hour. The product is first obtained as a yellow foam (Figure 3A). The yellow foam is shredded to a yellow powder and transferred to another 250-mL round-bottom flask. After drying under high vacuum overnight (<1.0 mmHg), the product is obtained as a yellow powder (containing diastereomers, 1:1 d.r., 6.71 g, 96 wt% purity, 67 yield). A second run provided a yellow powder (6.35 g, 88 wt%, 64% yield) (Figure 3B and C, Note 12 and 13).

Figure 2. (A) Silica gel packed flash chromatography column and crude product dissolved in CH2Cl2 (15 mL); (B) Loaded crude product on the silica gel column; (C) Column after elution with 200 mL of pentane and 300 mL pentane/EtOAc (5:1) (Photos provided by authors)
Figure 3. (A) Product 1 after drying under high vacuum (<0.1 mmHg) (foam); (B) and (C) Pure product 1 (Photos provided by checkers)
B. 3-Chloro-2-phenylpyridine (2). A 500-mL single-neck round-bottom flask equipped with a Teflon-coated magnetic stirring bar (2 cm x 0.5 cm) is charged with the dearomatized pyridine adduct (1, 7.18 g, 18.0 mmol, 1.0 equiv.), which is dissolved in 200 mL dry CH2Cl2 (Note 14) under ambient conditions. The mixture is cooled to 0 ℃ with an ice bath. N‐Chlorosuccinimide (2.89 g, 21.6 mmol, 1.2 equiv.) is dissolved in 50 mL dry CH2Cl2 and the solution is added dropwise to the reaction mixture over a period of 10 minutes using a dropping funnel (Figure 4A, Note 15). After the addition is complete, the reaction mixture is stirred at 0 ℃ for 1 h and afterwards the reaction temperature is allowed to raise to ambient conditions (23 ℃). Then, the ice bath and dropping funnel are removed and the reaction vessel is loosely plugged with a glass stopper. (Figure 4B). The reaction mixture is stirred overnight (16 h) under ambient conditions (Figure 4C). The reaction progress is monitored by thin layer chromatography (Note 16). After full consumption of the starting materials, the reaction flask is connected to a rotary evaporator and all volatiles are evaporated (50 ℃, 650 mm Hg, then 180 mm Hg) resulting in a brown oily residue (Figure 4D). This residue is dissolved in CH3CN (50 mL) (Note 4), and 6N HCl (200 mL) is added (Figure 4E, Note 17). The resulting mixture is stirred at 60 ℃ for 24 h (Figure 4F).

Figure 4. (A) Set-up of the reaction; (B) Reaction mixture after removal of the ice bath; (C) Reaction mixture after 16 h reaction time (D) Oily residue after evaporation of all volatiles; (E) Reaction mixture after addition of CH3CN and 6N HCl; (F) Reaction mixture after stirring at 60 ℃ for 1 min and 24 hours (Photos provided by authors)
After cooling to ambient temperature (23 ℃), the reaction mixture is transferred into a 1 L beaker and diluted with 100 mL distilled water (Figure 5A). Solid Na2CO3 (70 g) is carefully added to the mixture portion-wise (Note 18). When the CO2-formation subsided, the pH was measured to be 6 - 7 using universal indicator paper and the mixture is transferred to a 500 mL separatory funnel and extracted with CH2Cl2 (3 x 100 mL) (Figure 5B). The combined organic phases are dried over Na2SO4 (15 g) for 5 minutes (Note 19). The drying agent is removed by filtration through a funnel fitted with a filter paper (Whatman Grade 595, 150 mm) into a 500 mL flask and the filtration cake is washed with CH2Cl2 (3 x 10 mL) (Figure 5C). All volatiles are removed by rotary evaporation (50 ℃, 650 mm Hg, then 10 mm Hg), yielding the crude product as a brown oil (Figure 5D).

Figure 5. (A) Reaction mixture transferred into a 1 L-beaker; (B) Mixture transferred to a separatory funnel for liquid-liquid extraction; (C) Filtration set-up; (D) Residue of workup after evaporation of all volatiles (Photos provided by authors)
The crude product is subjected to flash column chromatography. A glass column (3 cm diameter) is wet-packed with silica gel (60 g) suspended in pentane to a total height of 19 cm (Figure 6A, Note 6 and 7). The crude product is dissolved in CH2Cl2 (5 mL) and loaded onto the silica gel (Figure 6B, Note 8). The flash column chromatography is run with a slight overpressure of argon. After the dissolved product is fully absorbed onto the silica, the silica gel filling is topped off with sea sand (3 cm) (Note 9). The column is sequentially eluted with 150 mL of pentane and 160 mL of eluent (pentane/EtOAc 15:1), which is discarded (Figure 6C, Note 10). Afterwards, the column is charged with 440 mL eluent (pentane/EtOAc 10:1) and the fractions are collected in 20 mL tubes, filled to 18 mL volume (Note 14). The fractions 8-21 are combined into a 1 L flask and all volatiles are removed by rotary evaporation (50 ℃, 650 mm Hg, then 180 mm Hg) (Note 20). The purified product is transferred to a 50 mL flask which is dried under high vacuum (<0.1 mmHg) overnight to give product 2 as a yellow oil (2.70 g, 94 wt% purity, 79% yield) (Figure 6D, Note 20). Rf = 0.5 (pentane/EtOAc = 10/1) (Note 21).

Figure 6. (A) Packed column and crude product dissolved in CH2Cl2; (B) Crude product loaded onto the silica gel; (C) Column after elution of 150 mL of pentane and 160 mL pentane/EtOAc (15:1). The collection is started at this point; (D) Product 2 after purification via flash column chromatography (Photos provided by authors)
Product 2 is further purified via bulb-to-bulb distillation under high vacuum (0.05 mmHg) at 160 ℃ (Figure 7A, Note 22). The product is collected from the bulb and obtained as a colorless oil (2.40 g, >99 wt% purity, 72% yield) (Figure 7B, Notes 23 and 24).
Figure 7. (A) Reduced pressure bulb-to-bulb distillation; (B) Pure 2 (Photos provided by checkers)
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/about/governance/committees/chemical-safety.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
2-phenylpyridine, methyl pyruvate, acetonitrile, dimethyl acetylenedicarboxylate, N-chlorosuccinimide, dichloromethane, hydrogen chloride, sodium carbonate, sodium sulfate, pentane, ethyl acetate, silica gel and chloroform.2.
2-Phenylpyridine (99.23%) was purchased from Ambeed and used as received.
3.
Methyl pyruvate (98%) was purchased from Thermo Scientific and used as received.
4.
Acetonitrile (>99.9%) was purchased from Fisher Chemical and used as received.
5.
Dimethyl acetylenedicarboxylate (98%) was purchased from Ambeed and used as received.
6. Silica gel "Silia Flash F60, 40-63 μm, 60 Å" was purchased from SiliCycle and used as received.
7.
Pentane (>98%) was purchased from Alfa Aesar and distilled before use.
8.
Dichloromethane (>99.5%) was purchased from Fisher Chemical and freshly distilled over
P2O5 before use.
9. Sea sand was purchased from EMD Millipore Corp. and used as received.
10.
Ethyl acetate (>99.5%) was purchased from Fisher Chemical and distilled before use.
11. TLC analyses were performed using
pentane/
EtOAc 2:1 as eluent and visualization was achieved with a 254 nm UV lamp. Aluminum backed TLC silica gel plates were purchased from SiliCycle. R
f-value of
2-phenylpyridine = 0.7, R
f-value of intermediate
1 = 0.5. After 36h, there is still starting material remaining (A). Fractions 4-12 are combined and evaporated to give intermediate A (B).
Figure 8. (A) TLC analysis of elution of the crude product with pentane/EtOAc (2:1) under visualization with UV lamp (λmax = 254 nm); (B) TLC analysis of collected fractions 4-12 (Photos provided by authors)
12. If the concentrated reaction mixture is evaporated under high vacuum overnight (16 h) before flash column chromatography product
1 can be obtained in improved 8.78 g, 88% yield.
13.
Trimethyl 2-methyl-6-phenyl-2H,9aH-pyrido[2,1-b][1,3]oxazine-2,3,4-tricarboxylate (
1) has the following properties: FTIR (neat):
ν (cm
-1) 2954, 1747, 1709, 1566, 1418, 1277, 1235, 1205, 1180, 1157, 1112, 1075, 1051, 976, 930, 766, 742, 730, 720, 696, 673.
1H NMR
pdf (400 MHz, CDCl
3): δ (ppm) 7.48 - 7.42 (m, 1H), 7.36 - 7.29 (m, 3H), 7.29 - 7.21 (m, 1H), 6.52 - 6.40 (m, 1H), 5.75 - 5.66 (m, 1H), 5.54 (d,
J = 4.3 Hz, 0.5H), 5.34 (d,
J = 6.1 Hz, 0.5H), 5.29 (d,
J = 6.1 Hz, 0.5H), 5.27 (d,
J = 4.4 Hz, 0.5H), 3.86 (s, 1.5H), 3.76 (s, 1.5H), 3.67 (s, 1.5H), 3.65 (s, 1.5H), 3.13 (s, 3H), 1.92 (s, 1.5H), 1.65 (s, 1.5H).
13C NMR
pdf (151 MHz, CDCl
3): δ (ppm) 171.1, 170.3, 165.9, 165.5, 163.2, 162.8, 141.9, 140.8, 139.7, 138.9, 136.3, 135.9, 128.8, 128.7, 128.4, 127.4, 126.8, 124.6, 123.5, 114.5, 113.7, 104.1, 104.0, 81.6, 80.7, 78.8, 78.2, 53.3, 52.9, 52.5, 52.3, 52.3, 24.7, 22.9. HRMS (ESI): calculated for C
21H
22NO
7: 400.139078; found: 400.141869. Purity of 96 wt% was determined by qNMR
pdf, using
1,3,5-trimethoxybezene as internal standard.
1,3,5-Trimethoxybezene (≥99%) was purchased from Sigma Aldrich and used as received.
14.
Dichloromethane (>99.5%) was purchased from Fisher Chemical and freshly distilled from P
2O
5 before use.
15.
N-Chlorosuccinimide (98.23%) was purchased from Ambeed and used as received.
16. TLC analyses were performed using
pentane/
EtOAc 7:3 as eluent and visualization was achieved with a 254 nm UV lamp. R
f-value of
1 = 0.4, R
f-value of product
2 = 0.5.
Figure 9. TLC analysis of the reaction mixture after 16 h reaction time under visualization with UV lamp (λmax = 254 nm) (Photo provided by authors)
17. 6N
HCl is obtained by diluting
HCl (37%) with equal volume of distilled water.
HCl (37%) was purchased from Fisher Chemical.
18.
Sodium carbonate (technical grade) was purchased from Spectrum Chemical MFG Corp. and used as received.
19.
Sodium sulfate (>99%) was purchased from Strem Chemicals and used as received.
20. TLC analyses were performed using
pentane/
EtOAc 9:1 as eluent and visualization was achieved with a 254 nm UV lamp. R
f-value of
2-phenylpyridine = 0.55, R
f-value of
2 = 0.5.
Figure 10. (A) TLC analysis after elution of the crude product with pentane/EtOAc 9:1 under visualization with UV lamp (λmax = 254 nm); (B) TLC analysis of collected fractions 8-21 (Photos provided by authors)
21. The purity of 94 wt% was determined by qNMR
pdf, using 1,3,5-trimethoxybenzene as internal standard.
22. Bulb-to-bulb distillation is conducted with a BÜCHI Glass Oven B-585.
23.
3-Chloro-2-phenylpyridine (
2) has the following properties: FTIR (neat):
ν (cm
-1) 3044, 1571, 1553, 1432, 1416, 1131, 1090, 1075, 1032, 1017, 794, 786, 738, 695, 681, 620, 612, 561, 532, 434.
1H NMR
pdf (400 MHz, CDCl
3) δ (ppm) 8.60 (dd,
J = 4.7, 1.5 Hz, 1H), 7.80 (dd,
J = 8.0, 1.5 Hz, 1H), 7.76 - 7.70 (m, 2H), 7.52 - 7.39 (m, 3H), 7.22 (dd,
J = 8.1, 4.6 Hz, 1H).
13C NMR
pdf (151 MHz, CDCl
3) δ (ppm) 156.7, 147.7, 138.4, 138.2, 130.3, 129.5, 129.0, 128.2, 123.2. HRMS (ESI): calculated for C
11H
9NCl: 190.041803; found: 190.041703. Purity of >99 wt% was determined by qNMR
pdf, using
1,3,5-trimethoxybenzene as internal standard.
24. On half scale,
3-chloro-2-phenylpyridine was obtained in 99% purity without bulb-to-bulb distillation as pale yellow oil (1.45 g, 85%).
pdf
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
Halopyridines are key intermediates en route to drug-related compounds,
2,3,4,5 but are also inherently valuable as bioactive compounds like for example etoricoxib. The most efficient methods to access these azines are pyridine C-H halogenation reactions. However, reported protocols for such transformations, especially for
meta-halogenation, are highly limited.
6,7,8,9 One of the exploited strategies rely on electrophilic aromatic substitutions of pyridines,
10,11 but those often lack efficiency due to the electronically mismatched nature of the processes due to the poor π nucleophilicity of pyridines.
2,3 These reactions normally require harsh reaction conditions, work only on a limited substrate scope and result often in poor regioselectivity control. Metalation-halogenation sequences with strong bases are other established approaches, but those require directing groups to functionalize the pyridine's
meta-position reliably.
12,13Recently, temporary dearomatization approaches have emerged as highly promising tools for
meta-functionalization of pyridines.
14 Within such strategies, the electron-deficient pyridines are first transformed into activated electron-rich enamine-type intermediates, which can then undergo electrophilic reactions, and those are subsequently rearomatized to finally provide
meta-substituted pyridines. Following this strategy, the McNally group developed a
meta-halogenation of pyridines
via a ring opening, functionalization, and ring-closing sequence through Zincke imine formation.
15 Meanwhile, our group introduced a redox-neutral dearomatization-rearomatization process for versatile
meta-halogenation of pyridines.
16 Off-note, along with halogenation also other functionalizations such as perfluoroalkylations, nitration, thiolation or selenylation can be achieved in the pyridine's
meta-position
via our strategy under either ionic or radical conditions. In our work, the azines are first dearomatized with commercial acetylenedicarboxylate (DMAD) and methyl pyruvate (MP) through a Huisgen 1,4-dipolar cycloaddition,
17,18 which generates the bench-stable, electron-rich oxazinopyridines (like
1) in excellent yields as diastereomeric mixtures. Along with pyridines, also quinolines, isoquinolines and thiazoles are readily dearomatized in good yields through this strategy. While the dearomatization and the following functionalization-rearomatization sequence can also be run as one-pot procedure, the isolation of the oxazinopyridines suppress potential side-reactions which may be caused by the remaining DMAD and MP.
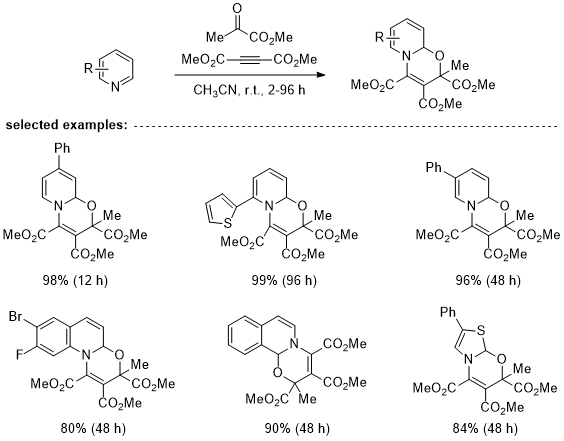
Table 1. Selected examples of dearomatized azines. For the full scope see reference
16.
The halogenations proceed either on the enamine's β- or δ-position of the oxazinopyridines through electrophilic substitution processes with the corresponding N-halosuccinimides as halide sources. Subsequent one-pot, acid promoted rearomatization provides the targeted halogenated pyridines with exclusive meta-selectivity in good to very good overall yields. Notably, mono-functionalized products are formed with high selectivity even though the respective oxazinopyridines formally possess two available meta-C-H positions, as the first halogenation significantly weakens the nucleophilicity of the halogenated enamine via their electron-withdrawing effect. Regarding the regioselectivity for 2-aryl pyridines, chlorination proceeds on the enamine's β‐position, while bromination and iodination is occurring on the δ-position. This may be attributed to the irreversibility of the chlorination due to the stronger nature of the C-Cl bond, which results in formation of the kinetic isomer (β-product). However, for bromination and iodination, initial halogenation might be reversible, and the subsequent deprotonation becomes the regioselectivity-determining step towards the exclusive formation of the thermodynamic product (δ-product). This method is also applicable for the 3-halogenation of quinolines, isoquinolines and thiazole compounds, as well as for halogenation of more sophisticated pyridine congeners such as drugs or natural products.
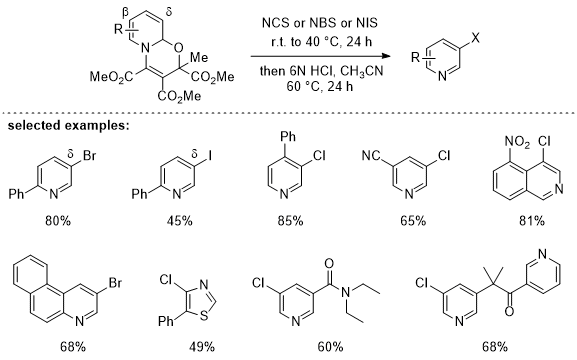
Table 2. Selected examples of the pyridine
meta-halogenation. For the full scope see reference
16.
In our publication, we also showed that the halogenation can be performed as one-pot processes without the isolation of the oxazinopyridine intermediates. Moreover, we demonstrated the selective sequential meta-meta'-dihalogenation without isolation of the mono-halogenated intermediate. These one-pot reactions further highlight the value of our method.
Table 3. Synthetic applications. For details see reference
16.
Appendix
Chemical Abstracts Nomenclature (Registry Number)
2-Phenylpyridine; (1008-89-5)
Methyl pyruvate; (600-22-6)
Dimethyl acetylenedicarboxylate; (762-42-5)
NCS: N-Chlorosuccinimide; (128-09-6)
Na2CO3: Sodium carbonate; (497-19-8)
Na2SO4: Sodium sulfate; (7757-82-6)

|
Pengwei Xu received his PhD in 2019 from East China Normal University under the direction of Prof. Jian Zhou. He then moved to the University of Hong Kong, working as a postdoctoral fellow with Dr. Zhongxing Huang. In 2022, he joined the group of Prof. Armido Studer as a Humboldt postdoctoral researcher, working on the method development for selective pyridine functionalization. |

|
Armido Studer received his PhD in 1995 (ETH Zürich, Dieter Seebach). He continued as a Postdoc at the University of Pittsburgh (Dennis P. Curran). In 1996 he started his independent career at the ETH. In 2000, he was appointed as Associate Professor in Marburg and in 2004 as Full Professor in Münster. His current research interests focus on the development of new synthetic methods, living radical polymerizations, the preparation of functional polymers, and the development of methods for the chemical modification of surfaces. |

|
Alexander Sailer studied chemistry at the universities of Erlangen, Würzburg, and Krakow. For his PhD, he joined the Thorn-Seshold group at LMU Munich, where he developed light-controllable inhibitors of the microtubule network. During this time, he was a visiting researcher in Dirk Trauner's group at New York University. In 2023, he returned to the Trauner group, now at the University of Pennsylvania, as a Humboldt Postdoctoral Fellow, focusing on the optical control of cAMP-dependent second messenger signaling. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved