Org. Synth. 2025, 102, 1-18
DOI: 10.15227/orgsyn.102.0001
Sharpless Epoxidation of Divinyl carbinol
Submitted by Klaus-Peter Rühmann
1, Paula Mora
1, and Dirk Trauner*
2Checked by Aaron L. Featherston, Nathan Ide, and Seble Wagaw
1. Procedure (Note 1)
A. (S)-1-((R)-oxiran-2-yl)prop-2-en-1-ol (2). A 1-L single necked round bottom flask (24/40 joint) is charged with a 4 cm Teflon-coated egg-shaped stir bar and powdered 4Å molecular sieves (10.0 g) (Note 2). The flask and a 250-mL graduated cylinder containing a 24/40 joint are dried overnight in a 160 ℃ oven. After 12 hours the flask and graduated cylinder are taken out of the oven and are sealed immediately with a rubber septum. Both systems are connected to high vacuum (≤0.1 mbar) by use of a needle adapter, connected to a Schlenk line and are allowed to cool to ambient temperature before refilling with an inert nitrogen atmosphere. Using the graduated cylinder, dichloromethane (240 mL, 0.5 M) (Note 3) is transferred by cannula to the reaction flask (Figure 1A). The round bottom flask containing the resulting white suspension is placed in an ice/salt bath (1:1, 800 g ice + 800 g salt) (Note 4) with an external temperature reading of the resulting slurry of -15 ℃ to -16 ℃ (Note 5). While stirring (300 rpm) (+)-diisopropyl L-tartrate (4.53 mL, 5.07g, 21.4 mmol, 0.18 equiv.) (Note 6) is added to the reaction flask (Figure 1B). Then freshly distilled titanium tetraisopropoxide (5.7 mL, 5.40g, 19.0 mmol, 0.16 equiv.) (Note 7) is added and the mixture stirred for 15 minutes followed by the dropwise addition of a cumene hydroperoxide solution (80wt%, 43.9 mL, 45.2g, 238 mmol, 2.00 equiv.) (Note 8,9) over the course of 5 minutes. The reaction mixture is stirred for an additional 15 minutes before the neat divinyl carbinol (11.6 mL, 10.0 g, 119 mmol, 1.00 equiv.) (Note 10) is added over the course of 3 minutes, turning the before colorless suspension deep orange (Figure 1C). The needle adapter is removed from the reaction flask, the septum is sealed with electrical tape and the flask is placed in a -20 ℃ freezer for 5 days (Figure 1D).

Figure 1: Reaction setup: A) Transfer of dichloromethane by cannula to the reaction flask, B) placement of reaction flaks into the ice/salt bath and addition of (+)-diisopropyl L-tartrate, C) Reaction mixture after complete addition of divinyl carbinol, D) Sealed septum with electrical tape and placement of the reaction flask in the -20 ℃ freezer. (photos provided by authors)
After 5 days the reaction flask is taken out of the freezer and the electrical tape and septum is removed. TLC analysis (
Note 11) indicated complete consumption of starting material (Figure 2A).
Acetone (200 mL) (
Note 12) and a solution of
citric acid (2.52 g) (
Note 13) in water (20 mL) (
Note 14) are added and the mixture is stirred for 1 h (350 rpm), while warming to ambient temperature (21 ℃). The reaction mixture is filtered over
celite (
Note 15) using a 9 cm diameter fritted Büchner funnel (Figure 2B) into a 2-L round bottom flask (Figure 2B). The reaction flask and
celite pad are washed with
dichloromethane (3 x 60 mL) and the filtrate is concentrated under reduced pressure using a rotary evaporator (100 mbar, 40 ℃ water bath) (
Note 16) affording approximately 70 g of a clear bronze colored crude oil (Figure 2C).
Figure 2: A) TLC-plate after staining with permanganate solution. Eluent: 40% ethyl acetate in hexanes. Lane 1: divinyl carbinol starting material. Lane 2: co-spot. Lane 3: reaction mixture. B) Filtration setup. C) Crude product after concentration. (photos provided by authors)
The crude product was purified by silica gel column chromatography (Notes 17,18,19 and 20), affording approximately 16 g of the desired product, contaminated with residual copolar diisopropyl tartrate as a colorless oil.
Figure 3: A) Dimensions for the column chromatography. B) TLC analysis of collected fractions. Fractions 13-35 were combined. (photos provided by authors)
Further purification can be achieved by fractional distillation. For this, the product obtained from column chromatography is transferred into a 50 mL round bottom flask, using
diethyl ether (2 x 10 mL) to wash remaining material from the larger flask. After concentration by rotary evaporation (100 mbar, 40 ℃ water bath), a 2 cm Teflon-coated egg-shaped stir bar was added to the round bottom flask and a short-path distillation set-up was assembled (Figure 4) (
Note 21). The product is distilled under vacuum and collected in fraction 2 (20 mbar, 70-72 ℃) (
Note 22) affording the pure epoxide
2 as a clear colorless liquid (7.53 g, 75.2 mmol, 63% yield, 96.9 wt% purity as determined by qNMR) (Figure 4B) (Notes
23,
24,
25, and
26). The enantiomeric excess (ee) was determined by chiral GC analysis and assessed to be >99% ee (Notes
29,
30).
Figure 4: A) Distillation set-up. B) Purified product. (photos provided by authors)
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/about/governance/committees/chemical-safety.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
1,4-pentadien-3-ol,
diisopropyl tartrate,
titanium(IV) isopropoxide,
tert-butyl hydroperoxide solution (5.5 M in decane), 4Å molecular sieves (powder),
dichloromethane,
acetone, deionized water,
citric acid,
celite, silica gel,
diethyl ether, hexanes,
1,3,5-trimethoxybenzene,
chloroform-d as well as the proper procedures for solvent evaporation and the application of high vacuum. Reactions and subsequent operations involving peracids and peroxy compounds should be run behind a safety shield. Peroxy compounds should be added to the organic material, never the reverse. New or unfamiliar reactions, particularly those run at elevated temperatures, should be run first on a small scale.
2. Powdered 4Å molecular sieves were purchased from Alfa Aesar
TM and used as received.
3.
Dichloromethane (99.8%, Extra Dry over Molecular Sieves, stabilized, with AcroSeal
®) was purchased from Acros Organics
TM and used as received.
4. Rock salt for the preparation of -20 ℃ cooling baths was purchased from Scotwood Industries, Inc. (de-icing salt). The checkers used
sodium chloride (ACS Reagent, >99.0%) as received from Sigma Aldrich.
5. The checkers used a PTFE coated needle tip thermocouple and monitored internal temperature at -15 ℃ to -20 ℃.
6. Both enantiomers
(+)-diisopropyl L-tartrate (98+%) and
(-)-diisopropyl D-tartrate (98+%) were purchased from Alfa Aesar
TM and used as received. Due to their high viscosities a 16-gauge needle is recommended for transfers. The checkers used
(+)-diisopropyl L-tartrate (98%, optical purity ee: 99%) and
(-)-diisopropyl D-tartrate (98%) as received from Sigma Aldrich.
7.
Titanium(IV) isopropoxide (98+%) was purchased from Acros Organics
TM and freshly distilled using a 10-mL Hickman still head before use. The checkers used fresh
titanium(IV) isopropoxide (97%) as received from Sigma Aldrich from single use 25mL bottles.
8. The authors used
tert-butyl hydroperoxide solution (~5.5 M in decane over 4Å molecular sieves) purchased from Sigma Aldrich and used as received.
9. Due to commercial availability, the checkers used
cumene hydroperoxide (technical grade, 80%) as received from Sigma Aldrich and obtained comparable results.
10.
1,4-pentadien-3-ol (98%, stabilized) was purchased from Acros Organics
TM and used as received. The checkers used
1,4-pentadien-3-ol (98%, stabilized with 0.4% hydroquinone) as received from Sigma Aldrich.
11. Glass support silica covered TLC plates (20 x 20 cm, 60G, F
254) were purchased from Sigma-Aldrich and were cut into 2.5 x 5 cm or 4 x 5 cm plates using a SiliaPlate TLC Plate Cutter (SiliCycle). A mini-workup refers to the following procedure: 500 uL of aqueous saturated
ammonium chloride solution and 100 uL of
diethyl ether were added to a 1 -mL vial. The reaction flask was opened and the tip of a Pasteur pipette was inserted first into the reaction mixture then into the 1 mL vial containing the biphasic mixture. The vial was capped and shaken before the organic layer was spotted on the TLC plate. The eluent used was 40%
ethyl acetate in hexanes. The TLC plate was stained by dipping into a
potassium permanganate solution (Dissolve 7.5 g of
KMnO4, 50 g
K2CO3, and 6.25 mL 10%
NaOH (or 8 mL, 1 M) in 1000 mL water. A typical lifetime for this stain is approximately 3 months.), followed by heating with a heatgun.
12. Reagent grade
acetone (ACS) was purchased from Fisher Scientific and was used a received. The checkers used
acetone (99.8%) from Sigma Aldrich as received.
13.
Citric acid (anhydrous) was purchased from Oakwood Chemical and used as received.
14. Deionized water was obtained and used directly from the university supply.
15. Celite
TM 545 was purchased from Acros Organics
TM and used as received. Procedure for the filtration over
celite: Add 40 g of
celite into a 9 cm diameter fritted Buchner funnel, followed by the addition of 150 mL of
dichloromethane. The slurry is stirred until homogeneous, then a vacuum is applied to remove DCM. Excess solvent was discarded before filtration of the reaction mixture.
16. The product is volatile. Despite measuring a boiling point of 72 ℃ at 20 mbar, significant amount of product was detected in the receiving flask of the rotary evaporator if pressures lower than 100 mbar or water bath temperatures higher than 40 ℃ were applied. In general, the evaporation time should be kept minimal and carefully monitored. The checkers performed the rotary evaporation at 30 ℃.
17. Silica Gel (Geduran® Si 60) for flash column chromatography (0.040-0.063 mm) was purchased from Sigma-Aldrich and used as received. Reagent grade
diethyl ether (BHT stabilized, ACS) was purchased from Fisher Scientific and used a received. Reagent grade hexanes (ACS, mixture of isomers) was from VWR and used a received. Dimensions of the column: 7 cm diameter, 30 cm height with a 1-L solvent reservoir.
18. When
tert-butyl hydroperoxide was used in the reaction, the authors used the following column chromatography procedure: 2 L of 10%
diethyl ether in hexanes was prepared. The column was packed as follows: 40 g of sand were added, followed by 200 mL of the 10% eluent to ensure an even top layer of sand. Next, the silica was wet-loaded (200 g of silica in 500 mL of the 10% eluent, followed by 2 x 100 mL rinses of the silica containing Erlenmeyer flask). Remaining silica on the inside of the column was washed down with another 100 mL of the 10% eluent. External pressure was created using an air pump. After complete elution of excess solvent another layer of sand (60 g) was added. The crude product was loaded onto the sand layer with 10 mL of the 10% eluent. The product containing round bottom flask was rinsed twice with 25 mL of the 10% eluent each. The remaining 940 mL of the 10 % eluent is added and collected as 2 fractions in 500 mL Erlenmeyer flasks. Elution is continued with 2 L of 50%
diethyl ether in hexanes and factions were collected in 50 mL test tubes (38 fractions). TLC analysis (40%
ethyl acetate in hexanes, stained with
potassium permanganate stain) indicated product in fractions 13-35. Product containing fractions were combined and concentrated under reduced pressure using a rotary evaporator (100 mbar, 40 ℃ water bath).
19. When
cumene hydroperoxide was used in the reaction, the checkers used a modified step gradient for the column chromatography as follows: 1 L of 15%
EtOAc/hexanes, 1L of 20%
EtOAc/hexanes and 40%
EtOAc/hexanes. The initial 500 mL eluate is collected in a 500-mL Erlenmeyer flask, and remaining fractions collected in 50 mL test tubes (49 fractions). TLC analysis (40%
ethyl acetate in hexanes, stained with Anisaldehyde stain) indicated product in fractions 10-49. Fractions 10-16 were contaminated with
2-phenyl-2-propanol and were discarded (Figure 5). Product fractions 17-49 were combined and concentrated under reduced pressure using a rotary evaporator (100 mbar, 40 ℃ water bath) to afford ~16 g of product contaminated with residual copolar
diisopropyl tartrate and
ethyl acetate. The checkers used
ethyl acetate (99.7%) and hexanes (98.5%) from Sigma Aldrich as received.
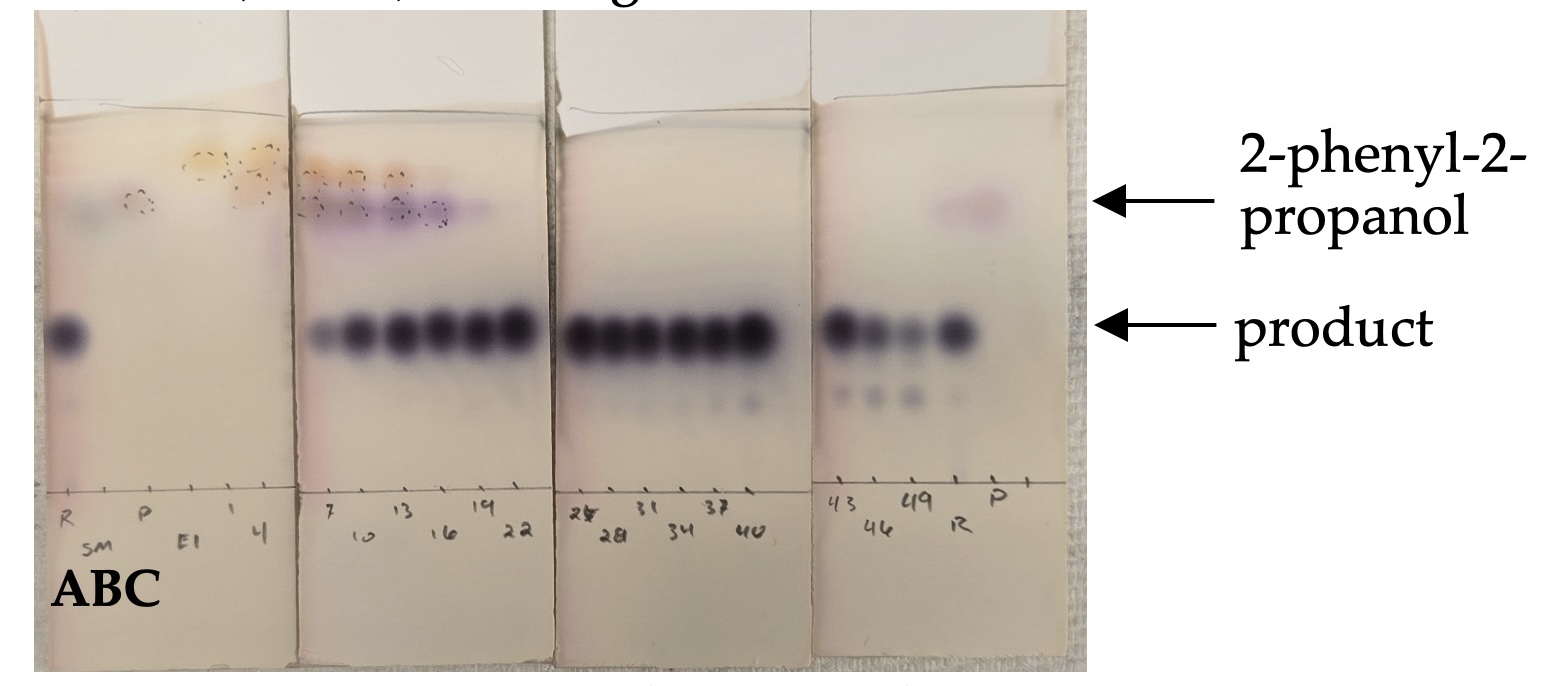
Figure 5: A) TLC-plate after staining with anisaldehyde stain. Eluent: 40% ethyl acetate in hexanes. Lane A: Crude reaction. Lane B: divinyl carbinol. Lane C: 2-phenyl-2-propanol. Product fractions 17-49 were combined. (Photo provided by checkers)
20. This initial silica plug was necessary to separate the
tert-butanol side product, or
2-phenyl-2-propanol in the case of
cumene hydroperoxide, from the crude mixture as it formed azeotropic mixtures with the product in the subsequent distillation step.
21. The distillation set-up was as follows: stir plate with heating block, 50-mL round bottom flask containing the product to be distilled with 2 cm Teflon-coated egg-shaped stir bar, short-path distillation bridge with thermometer for reading of the boiling temperature, the cooling jacket is connected to an ice/water (0 ℃) cooling cycle, the distill head to a vacuum pump (Büchi pump with accurate control of vacuum), and a three-way distillation spider connected to two pre-tared 10-mL (positions 1 and 3) and one tared 25-mL (position 2) receiving flasks.
22. Vacuum was applied slowly and at room temperature to prevent bumping of the compound. After vacuum was set to 20 mbar, the hot plate was slowly increased to 120 ℃. After 15 minutes, the boiling temperature rose from 21 ℃ to 72 ℃ and a forerun of 1 drop was collected. The receiving flask was switched to position 2 (25 mL flask) and the flask was submerged in an ice-bath. After an additional 20-25 minutes condensation ceased and the boiling temperature dropped below 60 ℃. The hot plate was slowly increased to 180 ℃, resulting in a rise of boiling temperature back to 72 ℃. After collection of a few more drops of product, the boiling temperature kept rising above 72 ℃ and the receiving flask was switched to position 3. Collection was stopped after the boiling temperature reached 86 ℃. Fraction 2 (7.53 g, 75.2 mmol, 63%) contained the pure product. Fraction 3 (393 mg) contained the product contaminated with
diisopropyl tartrate impurities.
23. Characterization data for (+)-
2: [α]
22D = +66.7° (c = 1.01, CHCl
3), bp = 70-72 ℃ at 20 mbar,
Rf = 0.26 (40%
ethyl acetate in hexanes, stained with
KMnO4),
1H NMR
pdf (400 MHz, CDCl
3) δ: 5.85 (ddd,
J = 17.0, 10.5, 6.3 Hz, 1H), 5.39 (dt,
J = 17.0, 1.4 Hz, 1H), 5.26 (dt,
J = 10.5, 1.4 Hz, 1H), 4.34 (dt,
J = 6.3, 2.7 Hz, 1H), 3.09 (dt,
J = 4.1, 3.0 Hz, 1H), 2.80 (dd,
J = 5.0, 2.8 Hz, 1H), 2.75 (dd,
J = 5.0, 4.1 Hz, 1H), 2.05 (d,
J = 2.7 Hz, 1H).
13C NMR
pdf (101 MHz, CDCl
3) δ: 135.6, 117.9, 70.3, 54.0, 43.6. IR (Diamond-ATR, neat): ṽ = 3417, 3081, 2996, 2927, 1645, 1427, 1251, 1132, 1104, 1069, 1026, 993, 930, 885, 793 cm
-1. HRMS (ESI): calculated for C
5H
9O
2 [M+H]
+: 101.0597, found: 101.0597.
24. The purity of product (+)-
2 was determined using qNMR
pdf on a sample prepared by dissolving 29.2 mg of (+)-
2 and 11.9 mg of
1,3,5-trimethoxybenzene (
Note 27) in 5 mL of
chloroform-d (
Note 28). The purity was determined to be 96.9 wt%
25. The authors report that different batches resulted in purities in a range of 96 wt% to 99.8 wt%.
26. In a second run on the same scale, the checkers obtained 6.78g (57% yield, 96.6 wt%) of (+)-
2 as a clear, pale yellow oil.
27. The internal NMR-standard
1,3,5-trimethoxybenzene was purchased from Santa Cruz Biotechnology (SCBT) and used as received. The checkers used
1,3,5-trimethoxybenzene (TraceCERT®) from Sigma Aldrich as received with a determined potency of 99.8 wt%.
28.
chloroform-d was purchased from Cambridge Isotope Labs and used as received. The checkers used
chloroform-d (99.8% d) from Sigma Aldrich as received.
29. Following the same procedure using
(-)-diisopropyl D-tartrate (
Note 5), (-)-
2 was obtained in 56.7% yield (6.75 g, 99.3 wt%, 99% ee) The optical rotation was determined to be [α]
22D = -67.6° (c = 1.01, CHCl
3). Chloroform (>99%) was obtained from Sigma Aldrich and used as received.
30. The enantiomeric excess was determined by chiral gas chromatography (GC)
pdf using a Shimadzu GC-2010 Plus, equipped with a chiral BETA DEX
TM 325 fused silica capillary column (30 m x 0.25 mm x 0.25 μm film thickness). The temperature gradient used was as follows: 3 minutes of equilibration at 25 ℃, followed by linear increase to 70 ℃ over 90 minutes (0.5 ℃/min); t
(+)-2 = 67.6 min, t
(-)-2 = 69.9 min. Samples were prepared by dissolving 2 uL of neat product in 1.5 mL
diethyl ether and an injection volume of 3 uL. A scalemic mixture was prepared by dissolving 6.29 mg of (+)-
2 and 7.52 mg of (-)-
2 in 5 mL
diethyl ether.
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
The fact that face-selective transformations on enantiotopic groups yield a single major product with exceptionally high enantiomeric excesses (ee) was first investigated and described by the Schreiber group in 1987.
3 These types of reactions combine an initial catalyst- or ligand-controlled selectivity for one of the four heterotopic faces with a subsequent kinetic resolution. An example they have already studied in detail in their initial publication is the transformation described in this Organic Syntheses procedure: the Sharpless epoxidation of divinyl carbinol (
1) (Scheme 1).
3,4 Divinyl carbinol has a mirror plane (σ), classifying it as an achiral starting material. The molecule has, however, two prochiral, enantiotopic vinyl groups that allow for desymmetrization. Upon exposure to asymmetric Sharpless epoxidation conditions the tartrate derived chirality within the transition state induces a face-selectivity favoring one major enantiomer of
2.
5 An initial selectivity of 88% ee was observed after only short reaction times. However prolonged exposure to the reaction conditions increased the enantiomeric excess due to a following kinetic resolution step.
4 This can be explained by the unreacted vinyl group of the undesired, minor enantiomer of
2 engaging in a second epoxidation reaction based on Sharpless' "matched" model, affording a bisepoxide of type
3. The desired enantiomer of
2 instead does not get epoxidized further as its unreacted olefin functionality is "mismatched" for a following second epoxidation. This enantiomer is enriched over time, ultimately affording an enantiopure (>99.9% ee) monoepoxide
2.
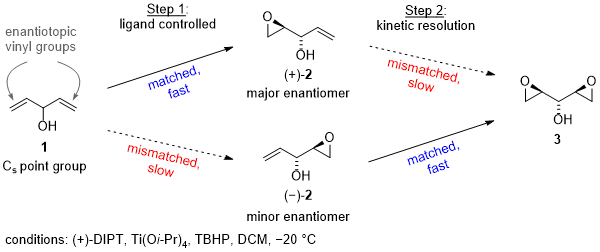
Scheme 1: The enantiotopos- and diastereoface-selective Sharpless epoxidation of divinyl carbinol (1) yields exceptionally high ee due to an initial ligand controlled facial selectivity followed by a kinetic resolution.
Figure 6. Examples of natural product total syntheses and approaches that utilized either enantiomer of epoxide 2 (highlighted in red)
6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23Practicality and versatility of monoepoxide
2 have been demonstrated in the total syntheses of a variety of natural products (Figure 6).
6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23 The different handles for functionalization allowed for the transformation of enantiomerically pure epoxides (+)-
2 or (-)-
2 into or towards a variety of natural products, including fatty acids, terpenes, alkaloids, polyketides, polyethers, oligosaccharides and others. Often the structural motif of
2 is deeply embedded within the natural product and its initial absolute configuration relays and induces selectivity for following transformations at later stages. Two elegant applications are showcased below.
In 1997 the research group of Paul A. Wender reported the total synthesis of resiniferatoxin (RTX) which is a potent TRPV1 agonist.
8,24 Isolated from
euphorbiaceae, this daphnane diterpene features an orthoester with a 5,7,6-fused tricyclic carbon skeleton.
25 Benzyl protection of the allylic alcohol in (-)-
2 was followed by treatment with Lewis acid and acetylide
4, affording the nucleophilic opening of the epoxide functionality with concomitant lactone formation (Scheme 2). Lactone
5 was converted to dihydropyranone
6 by use of an Achmatowicz reaction. Treatment of
6 with DBU then initiated the key (5+2) cycloaddition of the oxidopyrylium dipole in
7 with the alkene handle derived from epoxide
2, constructing the 6/7 bicyclic framework. The natural product was synthesized in a total of 43 steps.
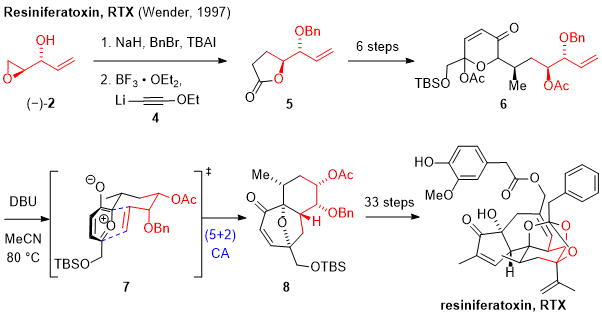
Scheme 2: Wender and coworker used the alkene functionality of (-)-2 within a (5+2) cycloaddition for the construction of the 6/7 bicyclic framework in the total synthesis of resiniferatoxin (RTX)
Loline alkaloids are a family of natural products that have been isolated from fescue grasses and show a wide range of biological activities.
26 In 2011 the Trauner group published an efficient access towards this natural product family, starting from epoxide (+)-
2 (Scheme 3).
15 Opening of the epoxide with 3-butenylamine was followed by a ring closing metathesis to afford the eight-membered heterocycle
10. The established (
Z)-alkene was then functionalized within a transannular aminobromination reaction, creating the pyrrolizidine core of the natural product. Four more steps were required to access loline.
Scheme 3: The alkene handle of epoxide (+)-2 was key in constructing the pyrrolizidine core of loline alkaloids in the efficient synthesis developed by Trauner and coworkers
Besides these two examples, many of the natural products listed in figure 6 show potent biological activities, such as anti-inflammatory, anti-bacterial, anti-cancer or immunosuppressant by different mechanisms of action.
Appendix
Chemical Abstracts Nomenclature (Registry Number)
4 Å Molecular sieves; 4 Å MS; (70955-01-0)
Dichloromethane; DCM; (75-09-2)
(+)-diisopropyl L-tartrate; (+)-DIPT; (2217-15-4)
(-)-diisopropyl D-tartrate; (-)-DIPT; (62961-64-2)
titanium tetraisopropoxide : titanium(IV) isopropoxide; Ti(Oi-Pr)4 (546-68-9)
tert-butyl hydroperoxide solution; TBHP; (75-91-2)
cumene hydroperoxide solution; (80-15-9)
divinyl carbinol: 1,4-pentadien-3-ol; (922-65-6)
citric acid; (77-92-9)

|
Klaus-Peter Rühmann obtained his Bachelor's and Master's degree at the Ludwig-Maximilian University of Munich, Germany in chemistry and biochemistry. He completed his Master's Thesis research in the group of Prof. Andrew G. Myers at Harvard University before he joined the graduate program at New York University in 2017 where he conducted synthetic studies towards different medicinally relevant natural products under the guidance of Prof. Dirk Trauner. He is currently a postdoctoral fellow with Jef Boeke at NYU Langone Health. |

|
Paula Mora graduated in chemistry from the University Complutense of Madrid (Spain) and conducted her Master's studies at the University of Birmingham (UK). In 2018 she started her PhD studies at the University of Vigo (Spain) under the supervision of Prof. Ángel R. de Lera and Prof. Rosana Álvarez. Her doctoral research is focused on the total synthesis of natural products as novel epigenetic modulators based on a bioinspired synthetic route. |

|
Dirk Trauner was born and raised in Linz, Austria, studied biology and chemistry at the University of Vienna, and received his undergraduate degree in chemistry from the Free University, Berlin. He then pursued graduate studies in chemistry under the direction of Prof. Johann Mulzer and became a postdoctoral fellow with Prof. Samuel J. Danishefsky at the Memorial Sloan-Kettering Cancer Center. Dr. Trauner joined the Department of Chemistry at the University of California, Berkeley, where he rose through the ranks to become an Associate Professor of chemistry (with tenure). In 2008, he moved to the University of Munich as a Professor of Chemistry and Chemical Genetics. In the Spring of 2017, he returned to the United States as the Janice Cutler Chair in Chemistry at New York University. In July 2022 he moved to the University of Pennsylvania as a Penn Integrates Knowledge Professor and George A. Weiss University Professor. |

|
Aaron Featherston is a Senior Scientist in the Process Chemistry group at AbbVie. He obtained his B.S. from Mercer University in 2014 and his Ph.D. from Yale University in 2020. At Yale, he worked on the development of peptide-based organocatalysts for asymmetric transformations with Professor Scott Miller. Following his graduate studies, Aaron joined the process team at AbbVie in September, 2020. |

|
Seble Wagaw joined Abbott in August 1999 in the Process Chemistry group in R&D. During her time at Abbott and AbbVie she has supported a wide range of development programs, including Viekira Pak and Maviret/Mavyret, AbbVie's first- and second-generation HCV cures. She was the Head of Process Chemistry from 2018 to 2022. Seble the served as the Vice President of the Small Molecule API group from 2022 to 2023, which consists of Process Chemistry, the Chemical Pilot Plant and the Program Management group. In August of 2023 Seble started her role as the Vice President of the CMC Strategy and Portfolio Leadership group. Seble holds a Ph.D. in Organic Chemistry from the Massachusetts Institute of Technology and a BS in Chemistry from the University of Michigan, Ann Arbor. Seble has two teenage children and in her spare time enjoys spending time with family, gardening, cooking (like all good chemists) and traveling. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved