Org. Synth. 2025, 102, 19-44
DOI: 10.15227/orgsyn.102.0019
Synthesis of Borane-Ammonia and its Application in the Titanium Tetrachloride Catalyzed Reduction of Carboxylic Acids, Including N-Protected Amino Acids
Submitted by Henry J. Hamann, Abdulkhaliq A. Alawaed, and P. Veeraraghavan Ramachandran*
1Checked by Gwyndaf A. Oliver, Konstantin Günther and Nuno Maulide.
1. Procedure (Note 1)
A. Borane-ammonia (1). A single-necked (29/42), air-dried, 2-L round bottom flask is charged with a Teflon-coated, egg-shaped magnetic stir bar (4.1 cm). Powdered sodium borohydride (NaBH4) (18.9 g, 500 mmol, 1 equiv) and powdered ammonium sulfate ((NH4)2SO4) (66.1 g, 500 mmol, 1 equiv) were weighed and added to the flask via powder funnel, open to air. (Notes 2 and 3). The flask was then cooled to 0 ℃ using an ice-water bath followed by the addition of 500 mL of reagent grade tetrahydrofuran (Figure 1a). The heterogeneous mixture is then vigorously stirred (1000 rpm) at 0 ℃ (Figure 1b), followed by the dropwise addition of water (4.5 mL, 250 mmol, 0.5 equiv) via glass pipette over a period of 5 min (Notes 4, 5, and 6, Figure 1c). Once the addition of water is complete, the flask is moved to room temperature (20-22 ℃). With continued vigorous stirring, tetrahydrofuran (20 mL) is added along the sides of the flask to wash the solids into the reaction mixture (Note 7).

Figure 1. a) Reaction solution for synthesis of 1; b) Reaction setup in ice bath for synthesis of 1; c) Reaction bubbling upon addition of water
The heterogeneous reaction mixture is stirred vigorously for 4-6 h at room temperature (20-22 ℃), and the flask was closed with a barbed hose adaptor connected to a vent line. (Notes 8, 9, and 10, Figure 2).
Figure 2. After stirring for 6 h at room temperature
Once complete, the contents of the reaction mixture are filtered under vacuum through a bed of celite (1-inch-thick) over a 500 mL sintered glass filter of coarse porosity (40-60 μm) into a 1-L filter flask (Figure 3a).
Figure 3. a) Filtration setup with Celite before filtration; b) Filtration setup showing filter cake following filtration
Residual solids within the reaction flask are dislodged using a spatula and additional tetrahydrofuran (3 x 50 mL) is added to the reaction flask to transfer the residue to the sintered glass filter. The filter cake is then washed with tetrahydrofuran (3 x 50 mL) to extract residual product from the filter cake (Figure 3b). The tetrahydrofuran extracts are concentrated to dryness in two portions by rotary evaporation in a 1-L, single-necked, evaporation flask (Note 11). The product is additionally dried under high vacuum (Note 12) for 12 h to obtain 1 (9.59 g, 62%) (Notes 13, 14, 15, and 16) as a white solid (Figure 4).
Figure 4. Sample of borane-ammonia (1)
B. tert-Butyl (2-hydroxyethyl)carbamate (3). A single-necked (24/40), air-dried, 250-mL round-bottomed flask is charged with a Teflon-coated, egg-shaped magnetic stir bar (2.6 cm). N-(tert-butoxycarbonyl)glycine (2) (3.50 g, 20.0 mmol, 1 equiv) is weighed and added to the flask open to air (Note 17). The flask is then charged with diethyl ether (20 mL, 1 M with respect to the acid) via a graduated cylinder, and the mixture is stirred to dissolve the carboxylic acid (Figure 5).
Figure 5. a) Reaction B setup; b) Close-up of reaction mixture
The flask is then sealed using a rubber septum and connected to an indirect inlet/outlet nitrogen line. The solution is cooled to 0 ℃ using an ice/water bath. With the reaction solution at 0 ℃, titanium tetrachloride (TiCl4) (0.33 mL, 3.0 mmol, 0.15 eq) is added dropwise to the stirred mixture through the septum using a syringe over a period of 5 min, resulting in a clear, yellow solution (Figure 6).
Figure 6. a) Reaction following TiCl4 addition; b) Close-up of reaction mixture
The septum is then opened and solid borane-ammonia (1.23 g, 40.0 mmol, 2 equiv) is added slowly, in 15 portions, to the stirred reaction mixture over a period of 25 min (Notes 18, 19, and 20). Upon complete addition of the borane-ammonia, the reaction flask is again sealed with a septum, and connected to an indirect nitrogen line. Following borane-ammonia addition the reaction is a white, heterogeneous mixture (Figure 7) (Note 21).
Figure 7. a) Reaction following borane-ammonia addition; b) Close-up of reaction mixture
After stirring at 0 ℃ for 15 min, at which point the reaction is a pale orange mixture, the reaction is removed from the ice/water bath and allowed to warm up to room temperature (20-22 ℃) (Figure 8).
Figure 8. a) Reaction after stirring for 15 min at 0 ℃; b) Close-up of reaction mixture
The reaction is stirred at room temperature (20-22 ℃) and monitored by 1H NMR spectroscopy until completion, typically 4 h (Note 22). On completion of the reaction, the crude mixture is brought to 0 ℃ using an ice bath and, open to air, is quenched by slow addition of cold 1 M HCl (30 mL) (Note 23) (Figure 9a). The mixture is brought to room temperature (20-22 ℃) and stirred for 10 min, then basified using 1 M NaOH (60 mL). Following basification, the reaction mixture is a pale blue to white, heterogeneous mixture (Note 24) (Figure 9b).
Figure 9. a) Reaction after acid addition; b) Reaction after base addition
The mixture is transferred to a 250-mL separatory funnel using water (10 mL) and diethyl ether (10 mL) to wash the reaction flask of residual product (Figure 10).
Figure 10. Reaction after transfer to separatory funnel
The organic layer is separated, and the aqueous fraction is added back to the separatory funnel and extracted with diethyl ether (4 × 50 mL) (Note 25). The combined organic layers are dried over anhydrous sodium sulfate (20 g), filtered through cotton, and concentrated under rotary evaporation (Note 26) (Figure 11).
Figure 11. Filtration setup
The remaining solvent is removed by stirring the mixture under high vacuum (Note 27) for 12 h to yield a clear, colorless liquid (3.04 g, 94%) (Note 28, 29, 30 and 31, Figure 12).
Figure 12. Sample of tert-Butyl (2-hydroxyethyl)carbamate (3)
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/about/governance/committees/chemical-safety.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with (
sodium borohydride,
ammonium sulfate,
tetrahydrofuran, celite,
N-(tert-butoxycarbonyl)glycine,
diethyl ether,
titanium tetrachloride,
borane-ammonia, and
sodium sulfate.
2. The following reagents were purchased from commercial sources and used without further purification by the authors:
sodium borohydride (powder, 98%, Oakwood Chemical), and
tetrahydrofuran (Sourced from Sigma-Aldrich, certified, contains 250 ppm BHT as inhibitor). Deionized water was used for the addition. The checkers used
sodium borohydride (granulated, >95%, TCI) which was ground using a mortar and pestle before use.
3.
Ammonium sulfate (colorless crystals, ACS grade 99%, Oakwood Chemical) was finely powdered prior to use (Figure 13), utilizing a mortar and pestle until no crystalline solid was visible (passed through 425-micron sieve). Decreased yields and longer reaction times will result if
ammonium sulfate is not powdered prior to use. The checkers used
ammonium sulfate (colorless crystals, ACS Reagent >99.0%, Sigma-Aldrich) which was ground to a fine powder, as described above. Grinding was found to be more effective when 10 smaller portions were ground separately before being combined. The checkers did not have access to a 425-micron sieve, but found that thorough grinding until no crystalline material was visible achieved the desired results in similar yields.
Figure 13. a) Powdering of ammonium sulfate; b) Granular ammonium sulfate; c) Powdered ammonium sulfate
4. The reaction should be carried out in a well-ventilated hood due to the hazards associated with the hydrogen gas released during the reaction.
5. The reaction is exothermic and an ice/water bath is needed for reactions conducted on this scale.
6. The addition of water leads to frothing due to the evolution of hydrogen gas. If there is appreciable froth formation, small portions of water can be added periodically over 15 min rather than continuous dropwise addition.
7. Periodically throughout the reaction,
tetrahydrofuran (20 mL) is added along the sides of the flask to wash the solids back into the reaction mixture.
8. The reaction progress is monitored using
11B NMR spectroscopy as judged by the absence of
sodium borohydride peaks (δ ≈ -43.6 ppm) in the
11B NMR spectrum. A reaction aliquot is withdrawn using a glass pipette and a drop of
DMSO is added to solubilize all of the
sodium borohydride prior to
11B NMR analysis.
11B NMR (96 MHz) δ -22.04 (q, J = 95.6 Hz).
9. Due to the highly heterogenous nature of the reaction, the reaction times can vary. Very vigorous stirring is essential for the reaction to be completed within the 4-6 h timeframe.
10. The reaction can alternatively be closed using a rubber septum attached to a vent through a needle.
11. Pressure: 40 mm Hg; Bath temperature: 35 ℃.
12. Pressure: 1 mm Hg; Temperature: room temperature (20-22 ℃).
13. A second run by the checkers, performed on half scale, gave 4.74 g (61%) yield. A run performed by the authors on full scale gave 10.11 g (66%).
14. Characterization data for
1: Colorless liquid.
1H NMR
pdf (600 MHz, Tetrahydorofuran-
d8) δ 4.24 - 3.80 (m, 3H, NH
3), 1.70 - 1.10 (m, 3H, BH
3);
11B NMR
pdf (193 MHz,
Tetrahydrofuran-
d8) δ -22.23 (q, J = 96.8 Hz). IR (ATR, thin film): 3308, 3250, 2376, 2279, 1598, 1373, 1155, 1055 cm
-1. The purity of the compound was determined to be >99% by hydride analysis
2, 3 (Figure 14).
Figure 14. Setup for hydride analysis of 1
15. Product
1 contains minor amounts of
2,6-di-tert-butyl-4-methylphenol (BHT) from the solvent
tetrahydrofuran. Using inhibitor-free
tetrahydrofuran provides
1 with no BHT contamination.
16. Recommended storage temperature: 2-8 ℃. Keep container tightly closed in a dry and well-ventilated location. Do not allow product to contact water during storage. Store separately from acids or oxidants. Samples of
1 have been stored without exclusion of air over a year without evidence of significant decomposition by
11B NMR spectroscopic analysis. The product should not be added to any waste containing acids or oxidants due to the potential formation of dihydrogen and other gases.
17. The following reagents were purchased from commercial sources and used without further purification:
N-(tert-butoxycarbonyl)glycine (crystalline powder, 98+%, Thermo Scientific),
diethyl ether (anhydrous, certified ACS, contains 5-10 ppm BHT as stabilizer), and
titanium tetrachloride (ReagentPlus, 99.9%, Sigma Aldrich).
18.
Borane-ammonia (
1) used was that prepared in step A.
19. The reaction should be carried out in a well-ventilated hood due to the hazards associated with hydrogen gas released during the reaction.
20. The addition of
borane-ammonia leads to frothing due to the evolution of hydrogen gas.
Borane-ammonia must be added in small portions to prevent over reaction.
21. The checkers found that the reaction mixture is prone to becoming an extremely viscous slurry at this stage unless the
borane-ammonia has been finely powdered prior to use.
22. The reaction progress is monitored using
1H NMR spectroscopy as judged by the absence of characteristic starting material (
N-(tert-butoxycarbonyl)glycine) peaks in the
1H NMR spectrum (
1H NMR (300 MHz, Chloroform-
d) δ 3.96 (d, J = 5.7 Hz, 1H)). A ~0.1 mL reaction aliquot is withdrawn using a glass pipette and the solvent removed with a stream of nitrogen. The resulting material is dissolved in chloroform-
d and subjected to
1H NMR analysis.
23. The addition of aqueous
HCl leads to frothing due to the evolution of hydrogen gas. Addition must be controlled and performed at 0 ℃ to prevent vigorous hydrogen evolution.
24. The resulting color of the quenched solution will vary from blue to white, depending on the exact quantities and rates of acid and base addition. The checkers observed in one case that the quenched solution became a deep blue color; the yield and purity of the obtained product was not affected.
25. If low yield of product is obtained, additional washes of the aqueous fractions may be required.
26. Pressure: 40 mm Hg; Bath temperature: 40 ℃.
27. Pressure: 1 mm Hg; Temperature: room temperature.
28. A second run by the checkers, performed on half scale, gave 1.33 g (83%) yield. A run performed by the authors on full scale gave 2.75 g (85%).
29. Characterization data for
3:
1H NMR
pdf (700 MHz, Chloroform-
d) δ 5.02 (s, 1H), 3.68 (t,
J = 5.1 Hz, 2H), 3.27 (q,
J = 5.5 Hz, 2H), 2.71 (s, 1H), 1.43 (s, 9H);
13C NMR
pdf (176 MHz, Chloroform-
d) δ 157.0, 79.8, 62.7, 43.3, 28.5. IR (ATR, thin film): 334, 2976, 2932, 1682, 1517, 1454, 1392, 1365, 1275, 1249, 1165, 1064 cm
-1. HRMS (ESI+) m/z calc'd for C
7H
15NNaO
3+ [M + Na]
+: m/z 184.09441, found: 184.0943. The purity of the compound was determined to be 98% by quantitative
1H NMR
pdf analysis using 11.1 mg of dimethyl fumarate (purity: 98%) as the standard and 14.6 mg of
3. The signal from
3 was selected (3.27) and compared against the signal from dimethyl fumarate (6.85) for calculations.
30. A reaction substituting methyl
tert-butyl ether (MTBE) for
diethyl ether provided 2.38 g (74%) of the product.
31. Recommended storage conditions: The product is chemically stable under standard ambient (room temperature) conditions. Keep container tightly closed in a dry and well-ventilated location. Store separately from strong oxidants. The product should not be added to any waste containing strong oxidants.
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
The reduction of carboxylic acids to the corresponding alcohols is a fundamental transformation in organic chemistry. The use of hydride reducing agents is made difficult by the weak electrophilicity
4 of the carbonyl carbon; despite this many such reagents are frequently used to achieve this important, yet challenging, transformation. Commonly utilized aluminum hydride reagents including lithium aluminum hydride (LiAlH
4),
5, 6 alane (AlH
3),
7, 8 and diisobutylaluminum hydride (DIBAL-H)
9 pose significant hazards due to their pyrophoricity,
10 extreme moisture sensitivity, and explosive incompatibility with certain functional groups.
11 Boron hydride alternatives such as borane-dimethyl sulfide (BMS)
12, 13, 14 which is efficient only for the reduction of aliphatic acids, and borane-tetrahydrofuran (BTHF) which is effective for reduction of aromatic or aliphatic acids also suffer from moisture sensitivity in addition to the long term instability of BTHF and the malodorous dimethyl sulfide byproduct of BMS.
Alternative methods of carboxylic acid reduction include the use of other metal hydrides,
15 metal borohydrides activated by additives,
16, 17, 18, 19, 20, 21, 22, 23, 24 direct hydrogenation with H
2 using a wide variety of catalysts,
25 reduction using silane reagents,
26, 27 and electron transfer or other radical reactions using SmI
2 or other reagents.
28, 29 The drawbacks of these methods are the required high temperature and pressure,
30, 31, 32, 33, 34 cost of the reagents, safety concerns,
18, 19, 35, 36 as well as undesirable side reactions including esterification of the carboxylic acid starting material with the product alcohol.
30 These issues warrant continued exploration of alternate approaches to carboxylic acid reduction.
Lewis acid-base adduct borane-amines, first synthesized over eight decades ago,
37 have also been explored as alternative reagents for carboxylic acid reduction. Borane-amines, including borane-ammonia, are easily prepared,
38, 39, 40 air- and moisture stable, and are generally considered as safer alternatives to traditional borane reagents. Their safety and stability are due to the increased donor ability of the amine nitrogen relative to oxygen and sulfur adducts with borane. However, this increased stability often renders borane-amines as weak reducing agents, necessitating higher reaction temperatures in reactions using relatively stable borane-amines such as borane-pyridine
41 and borane-triethylamine.
42 Less stable substitutes such as borane-
N,
N-diethylaniline
43 and borane-
N-ethyl-
N-isopropylaniline-borane
44 are subject to decomposition, leading to limited substrate scope and poor product yields.
The prototypical borane-amine, borane-ammonia, widely used as a hydrogen source
45, 46 for energy applications
47 and transfer hydrogenation reactions,
48 has also been utilized in wide range of functional group transformations.
49, 50 Recently we have reported several titanium tetrachloride (TiCl
4) accelerated reaction using borane-ammonia including the deoxygenation of esters to ethers,
51 the deoxyhalogenation of carbonyl compounds,
52 and the reductions of ketones,
53 carboxylic acids,
54 nitriles,
55 and amides.
56Our investigation of TiCl
4 accelerated reduction of carboxylic acids using borane-ammonia began following our related ketone reduction.
53 During the ketone reduction project it was observed that the substrate 4-acetylbenzoic acid provided a minor amount of 1-(4-(hydroxymethyl)phenyl)ethan-1-ol byproduct along with the expected 4-(1-hydroxyethyl)benzoic acid product as result of the concurrent ketone and acid reduction. Following up this observation, we pursued the application of the TiCl
4/borane-ammonia system to carboxylic acids.
54 An initial concern was the potentially competing amidation reaction to form benzamide as a product, which we have previously reported.
57 Delightfully, however, following a reaction applying the ketone reduction conditions to representative 3-phenlypropanoic acid, and 76% yield of the corresponding 3-phenylpropanol was obtained, without any byproduct amidation detected. Following investigation of reagent equivalency, catalyst loading, reaction solvent, alternative catalysts, and alternative borane-amines, we arrived at the optimal reaction conditions, 10 mol% TiCl
4, 2 equiv borane-ammonia, in diethyl ether (0.33 M) at room temperature.
Using the optimized conditions, a variety of aromatic and aliphatic carboxylic acids underwent conversion to the corresponding alcohols (Table 1). Substrates containing potentially reactive functionalities including halogen, α-halo, nitro, alkenes, and hydroxyl were well tolerated. Competitive reactions showed preferential reduction of ketones over the carboxylic acid, but moderate selectivity towards carboxylic acid reduction versus esters and amides. Selectivity was also demonstrated towards the reduction of aliphatic acids over aromatic acids. Completely selective reduction of carboxylic acid over nitrile functionality was further observed.
54 A large-scale (100 mmol) reduction was performed for phenylacetic acid, where a slightly decreased yield was observed using the conditions optimized at 1 mmol, however, upon increasing the TiCl
4, loading to 15 mol%, an isolated yield of 94.7% was obtained. Other minor modifications were necessary for the reduction of benzoic acid, which required 12 h reaction time, and the reduction of benzoylformic acid, which necessitated an increase to 3 equiv borane-ammonia and 50 mol% TiCl
4 due to the presence of multiple reducible functional groups.
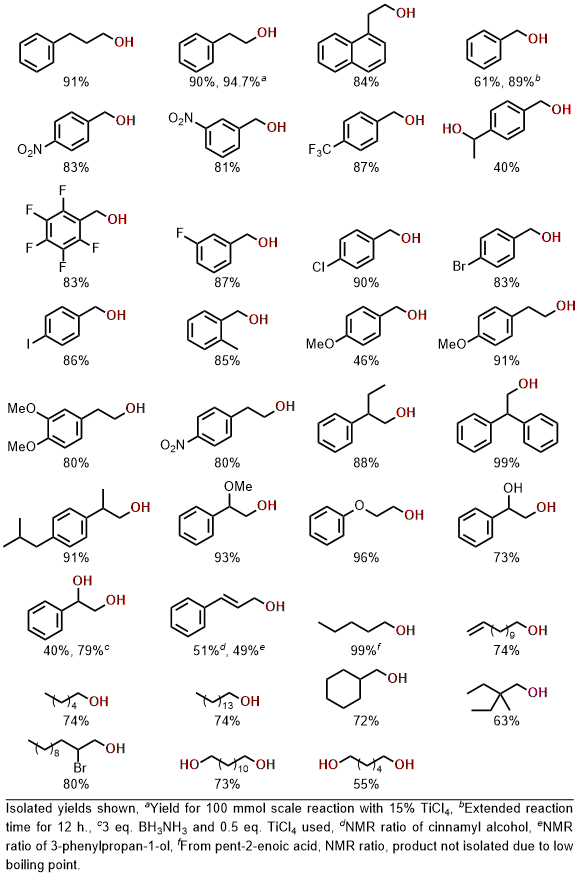
Table 1. Substrate scope for aliphatic and aromatic carboxylic acids
Having devised a broadly applicable reduction of aliphatic and aromatic carboxylic acids, substrates bearing amine functionalities were investigated. Unprotected aromatic and aliphatic amines, unfortunately, were not well tolerated, providing 38% and 22% of the corresponding alcohols respectively. However, amino acids with tert-butyloxycarbonyl (Boc) or fluorenylmethoxycarbonyl (Fmoc) protecting groups on the amine provided 74% to 93% of the corresponding alcohol products without cleavage of the protecting group. Complete retention of substrate stereochemistry was demonstrated by the reduction, deprotection, derivatization using menthyl chloroformate, and analysis using gas chromatography for Fmoc-valine.
Table 2. Substrate scope for N-protected amino acids
Thus, our research group has described a methodology for the TiCl4 promoted reduction of carboxylic acids using borane-ammonia under mild conditions. The described procedure provides a representative example of this methodology while demonstrating the tolerance of carboxylic acid substrates containing protected amines. The methodology can be used to reduce a wide variety of carboxylic acids, one of the fundamental reactions of organic chemistry, and provides an attractive alternative to currently employed reduction methodologies.
Appendix
Chemical Abstracts Nomenclature (Registry Number)
Sodium Borohydride: Borate(1-), tetrahydro-, sodium (1:1); (16940-66-2)
Ammonium sulfate: Sulfuric acid ammonium salt (1:2); (7783-20-2)
Water: Water; (7732-18-5)
Tetrahydrofuran: Furan, tetrahydro-; (109-99-9)
Borane-ammonia: Boron, amminetrihydro-, (T-4)-; (1) (Borane-ammonia)
N-(tert-Butoxycarbonyl)glycine: N-(tert-Butoxycarbonyl)glycine; (2) (4530-20-5)
Titanium tetrachloride: Titanium chloride (TiCl4) (T-4)-; (7550-45-0)
Diethyl ether: Ethane, 1,1'-oxybis-; (60-29-7)
tert-Butyl (2-hydroxyethyl)carbamate: Carbamic acid, N-(2-hydroxyethyl)-, 1,1-dimethylethyl ester; (3) (26690-80-2)

|
Dr. Henry J. Hamann obtained his B.A. in Industrial Design and his B.S. in Chemistry from Purdue University in 2012 and 2015 respectively. He received his PhD under the guidance of Professor P. V. Ramachandran at Purdue University in 2021. His research focuses on developing methodologies pertaining to the synthesis of borane-amines and their applications for organic synthesis and nanomaterials. |

|
Abdulkhaliq Alawaed finished his bachelor's degree in chemistry at Buffalo University in 2014, followed by the completion of his master's degree in organic chemistry from Oswego University in 2016. In 2019, he made the move to Purdue University and joined the research group led by P.V. Ramchandran. His ongoing doctoral research is centered on Lewis acid-catalyzed functional group transformations, with a particular emphasis on reactions involving borane-ammonia. |

|
Prof. P. V. Ramachandran received his B. Sc. and M. Sc. from the University of Calicut, India and his Ph. D. from the Indian Institute of Technology, Kanpur under the tutelage of Prof. Subramania Ranganathan. Subsequently he joined the laboratories of Prof. Herbert C. Brown at Purdue University as a postdoctoral fellow. He began his academic career at Purdue in 1997 where he is currently a Professor of Chemistry. His research interests are in the areas of synthetic methodologies, materials chemistry, and drug discovery. |

|
Gwyndaf A. Oliver received his MChem in 2019 from Magdalen College, University of Oxford, undertaking his Masters research with Prof. Michael C. Willis developing methodologies with SO2 surrogates. In 2023, he completed his PhD in the group of Prof. Daniel B. Werz at the University of Freiburg, on synthetic methodology using donor-acceptor cyclopropanes. Currently, he is a postdoctoral researcher in Prof. Nuno Maulide's group at the University of Vienna. His research interests span a wide range synthetic methodologies including organosulfur chemistry and reactive intermediates. |

|
Konstantin Günther studied chemistry at the Humboldt University of Berlin. After a Bachelor thesis on photomagnetic molecular switches with Prof. Oliver Dumele, he then joined the group of Prof. Ken'ichirō Itami at Nagoya University for his Master thesis on the synthesis of water-soluble aromatic nanobelts. Currently, he is a graduate student at the University of Vienna and the Center for Molecular Medicine (CeMM). Under the supervision of Prof. Nuno Maulide, he conducts research on the synthesis and target identification of novel macrocyclic immunosuppressants. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved