Org. Synth. 2025, 102, 64-85
DOI: 10.15227/orgsyn.102.0064
A General Method for C-Alkylation of Nitroalkanes with Alkyl Halides: Nickel Photoredox Dual-Catalyzed C-Alkylation of Nitroalkanes
Submitted by Sina Rezazadeh, Allyssa M. Conner, and Donald A. Watson*
1Checked by Neetu Sharma and M. Kevin Brown
1. Procedure (Note 1)
A. 2-(3-Iodopropyl)isoindoline-1,3-dione (1). A 250-mL one-necked (29/42 joint) round-bottomed flask equipped with a Teflon-coated magnetic stir bar (oval, 25 mm x 12 mm) was charged with triphenylphosphine (6.29 g, 24.0 mmol, 1.2 equiv) (Note 2), Imidazole (1.63 g, 24.0 mmol, 1.2 equiv) (Note 3), and dichloromethane (CH2Cl2) (40 mL) (Note 4). The flask was placed in a water/ice bath. The mixture was stirred (600 rpm) for 5 min under air atmosphere. Iodine (6.09 g, 24.0 mmol, 1.2 equiv) (Note 5) was then added to the mixture all at once while stirring. The sides of the flask were washed with CH2Cl2 (10 mL). The mixture was stirred for 15 min, during which an orange/brown suspension formed. 2-(3-hydroxypropyl)isoindoline-1,3-dione (4.10 g, 20.0 mmol, 1.0 equiv) (Note 6) was then added to the flask all at once. The sides of the flask were washed with CH2Cl2 (10 mL) and a rubber septum was placed on top of the flask to prevent solvent evaporation. The water/ice bath was removed and the mixture was allowed to warm up to room temperature (20 ℃) and stirred for 12 h. The septum was then removed and the mixture was poured into a 250-mL separating funnel. The flask was rinsed with additional CH2Cl2 (3 x 10 mL) and the washings were added to the separating funnel. Next, deionized water (75 mL) was added to the separating funnel, and the funnel was sealed with a cap. The funnel was shaken for efficient mixing of the layers and then vented briefly. The cap was removed, and the mixture was left standing, uncapped, until complete separation of the phases. The organic (bottom) layer was collected into a 250-mL Erlenmeyer flask. The aqueous phase was similarly washed with dichloromethane (2 x 25 mL). The combined organic phase was dried over anhydrous magnesium sulfate (≈4 g) (Note 7). A medium porosity sintered glass funnel (6.5 cm OD x 6 cm height) with a vacuum adapter was connected to a 500-mL single-necked (29/42 joint) round-bottom flask and a mild vacuum (≈50 mm Hg) was applied. The dried organic phase was filtered through the funnel and collected into the round-bottom flask. The Erlenmeyer flask was rinsed with additional CH2Cl2 (2 x 10 mL) and the washings were filtered through the glass funnel and collected into the round-bottom flask. The sides of the sintered glass funnel were also washed with CH2Cl2 (10 mL), which was collected into the round-bottom flask. To prepare the crude material for purification, silica gel (25 g) (Note 8) was added to the flask. The mixture was concentrated under reduced pressure on a rotary evaporator (30 ℃, 12 mm Hg) (Note 9) to leave a fine powder of silica gel with the adsorbed crude material. This material was charged on a column (7 cm OD x 30 cm height) of dry silica gel (100 g) and eluted with 200 mL of 75:25 hexanes/dichloromethane, and then 1100 mL of 50:25:25 hexanes/dichloromethane/ethyl acetate (Note 10, 11, 12). The eluent was collected over 20 fractions (65 mL glass tubes), with the desired product obtained in fractions 8 - 13. The fractions containing the product were combined and concentrated under reduced pressure on a rotary evaporator (30 ℃, 12 mm Hg). The obtained solids were transferred to a 20-mL scintillation vial and dried on a vacuum manifold (20 ℃, 0.05 mm Hg) overnight to provide the desired product (1) as a bench stable, white solid (6.08 g, 96% yield, 99.9 % purity) (Note 13, 14).

Figure 1. Left: triphenylphosphine and imidazole are added to the flask; Middle: reaction turns orange/brown after addition of iodine; Right: the mixture is stirred at room temperature after the addition of 2-(3-hydroxypropyl)isoindoline-1,3-dione (Photos are provided by the authors)
Figure 2. Left: crude mixture is washed with water; Middle: MgSO4 is filtered; Right: product (1) as a white solid (Photos provided by the authors).
B. 2-(3-(1-Nitrocyclohexyl)propyl)isoindoline-1,3-dione (2). A 1-dram (3.7 mL) glass vial equipped with a Teflon-coated magnetic stir bar (oval, 7 mm x 3 mm) was charged with 2,9-dimethyl-4,7-diphenyl-1,10-phenanthroline (bathocuproine) (180 mg, 0.5 mmol, 0.1 equiv) (Note 15) under air at RT. Nickel (II) bromide 2-methoxyethyl ether complex (NiBr2·diglyme) (176 mg, 0.5 mmol, 0.1 equiv) (Note 16) was quickly (Note 17) weighed on a weighing paper (Note 18) and added to the vial. Dioxane (1 mL) (Note 19) was added via a glass pipet to wash down any leftover solids on the weighing paper and the sides of the vial. The vial was sealed with a Teflon-coated polypropylene cap (Note 20) and the mixture stirred (1000 rpm) at room temperature (20 ℃) for 1 h during which a pink suspension formed.
A 20-mL scintillation vial was equipped with a Teflon-coated magnetic stir bar (oval, 20 mm x 10 mm). Potassium tert-butoxide (785 mg, 7.0 mmol, 1.4 equiv) (Note 21) was quickly (Note 22) weighed on a weighing paper and added to the vial under air. Dioxane (8 mL) was then added and the vial was sealed with a Teflon-coated polypropylene cap (Note 23). The mixture was stirred (1000 rpm) at room temperature (20 ℃) for 5 min (Note 24) after which a cloudy suspension formed. Nitrocyclohexane (969 mg, 7.5 mmol, 1.5 equiv) (Note 25, 26) was weighed in a 1-mL disposable syringe equipped with a 22-gauge x 4'' needle. While stirring, the cap of the 20-mL vial was removed and nitrocyclohexane was added drop-wise over 1 min to the mixture during which a white suspension formed (a slight exotherm was observed). The cap was replaced and the suspension was stirred vigorously (1000 rpm) for 10 min. Tris[2-phenylpyridinato-C2,N]iridium(III) (Ir(ppy)3) (8.2 mg, 0.01 mmol) (Note 27) and 2-(3-iodopropyl)isoindoline-1,3-dione (1) (1.58 g, 5.0 mmol, 1.0 equiv) were weighed on a weighing paper. The vial was removed from the stirrer, the cap of the vial was removed, and the solids were added all at once. The nickel complex, formed as a suspension in the 1-dram vial, was transferred via a glass pipet to the reaction mixture under air. (Note 28) The 1-dram vial was further washed with dioxane (1 mL), which was added to the reaction mixture via the pipet. The cap was then replaced and the mixture was shaken vigorously for 1 min, during which a green suspension with yellow solids formed (Figure 3, middle left). Next, the vial was placed in a Penn PhD Photoreactor m2 with the following set up: stir rate: 1000 rpm, fan rate: 6800 rpm, LED power: 100%, time: 12 h. (Note 29, 30, 31).

Figure 3. Top : A. suspension of NiBr2·bathocuproine; B. solution of Potassium tert butoxide in dioxane; C. suspension of potassium nitronate in dioxane; D. the reaction suspension after shaking for one min; Bottom: E. setup with photoreactor off; F. setup with photoreactor on; G. reaction mixture after completion as a green suspension with yellow solids (Photos E and F are provided by the checkers and all other photos are provided by the authors)
After 12 h, the reaction was removed from the photoreactor. The cap was then removed and ethyl acetate (8 mL) was added all at once to the green-yellow suspension under air. A medium porosity sintered glass funnel (3.5 cm OD x 5 cm height) with a vacuum adapter was connected to a 250-mL one-necked (24/40 joint) round-bottom flask. The glass funnel was loaded with Celite (≈1.5 cm height) (Note 32), which was moistened with ethyl acetate. A mild vacuum (≈50 mm Hg) was applied, and the suspension was passed through Celite to remove the solids. The vial was further rinsed with ethyl acetate (3 x 10 mL), which was passed through Celite. The glass funnel and Celite were also washed with ethyl acetate (2 x 10 mL). The collected green solution (Figure 4, left) was concentrated under reduced pressure on a rotary evaporator (35 ℃, 12 mm Hg) to give the crude material as a green oil. The crude material was diluted with dichloromethane (2 mL) and charged on a column (4.5 cm OD x 30 cm height) of silica gel (80 g) wetted with hexanes. The flask was washed with additional dichloromethane (2 x 2 mL), which was charged on the column. The column was eluted with 50 mL hexanes, then 50 mL of 95:5 hexanes/ethyl acetate, then 200 mL of 90:10 hexanes/ethyl acetate, then 400 mL of 85:15 hexanes/ethyl acetate, and finally 400 mL of 80:20 hexanes/ethyl acetate (Note 33). The eluent was collected in 51 fractions (22 mL glass tubes), with the desired product obtained in fractions 36-47. The fractions containing the product were combined and concentrated under reduced pressure on a rotary evaporator (30 ℃, 12 mm Hg) to give an off-white solid. The solid was transferred to a 20 mL scintillation vial and hexanes (3 mL) and ethyl acetate (2 mL) were added to the vial under air. The vial was heated on a 250 ℃ hot plate with continuous swirling until most of the solids were dissolved. Then additional ethyl acetate (0.2-0.3 mL) was added with heating to obtain a clear solution. The solution was left on the bench for 10 min to cool down to room temperature. The vial was sealed with a polypropylene cap and left at room temperature for an additional hour, after which it was placed in a freezer (-25 ℃) overnight. The supernatant was decanted from the white crystals with a 5" glass pipet. The crystals were swirled with hexanes (2 mL), which was decanted similarly with the pipet. The dried crystals were crushed with a spatula to a fine powder and further dried on a vacuum manifold (20 ℃, 0.05 mm Hg,) overnight to yield the desired product (2) as a bench stable, white crystalline solid 1.14 g (72 % yield) of product (2) in 99.4 % purity. (Note 34, 35).

Figure 4. Left: the crude material is filtered through Celite to leave a green solution; Right: pure product (2) as white crystals (Photos are provided by the authors)
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/about/governance/committees/chemical-safety.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
2,9-dimethyl-4,7-diphenyl-1,10-phenanthroline,
nickel(II) bromide 2-methoxyethyl ether complex,
dioxane,
hexanes,
diethyl ether,
triphenylphosphine,
imidazole,
iodine,
dichloromethane,
2-(3-hydroxypropyl)isoindoline-1,3-dione,
magnesium sulfate,
silica gel,
ethyl acetate,
Potassium tert-butoxide,
nitrocyclohexane,
tris[2-phenylpyridinato-C2,N]iridium(III), and
Celite. High intensity blue LED light can be damaging to eyesight. It is advised to cover the surroundings of the reaction setup and use blue-light blocking safety glasses to minimize the light exposure and protect the researchers.
2.
Triphenylphosphine (99%) was purchased from Sigma Aldrich and used as received.
3.
Imidazole (99+%) was purchased from Neta-Oakwood and used as received.
4.
Dichloromethane (Stabilized/Certified ACS) was purchased from Fisher Scientific and used as received.
5.
Iodine (99.8%) was purchased from Sigma Aldrich and used as received.
6.
2-(3-hydroxypropyl)isoindoline-1,3-dione (98%) was purchased from Combi-Blocks and used as received.
7. Anhydrous
magnesium sulfate was purchased from Oakwood Chemical and used as received.
8. SiliaFlash F60 (40-63 μm)
silica gel was purchased from Silicycle Inc. and used as received.
9. A Büchi Rotavapor R-124 rotary evaporator equipped with a Chemglass CG-4812-30 vacuum pump was used.
10.
Ethyl acetate (Certified ACS) was purchased from Fisher Scientific and used as received.
11.
Hexanes (Certified ACS) was purchased from Fisher Scientific and used as received.
12. The course of the chromatography can be monitored by TLC (EMD Millipore, TLC
silica gel 60 F
254). Product (
1): Rf = 0.43, 80:20
hexanes/EtOAc, visualized with a UV lamp (254 nm).
2-(3-hydroxypropyl)isoindoline-1,3-dione: Rf = 0.45, 40:60
hexanes/EtOAc, visualized with a UV lamp (254 nm).
Figure 5. Left: TLC of crude reaction and starting materials (80:20 hexanes/EtOAc as eluent); Right: TLC of crude reaction and starting materials (40:60 hexanes/EtOAc as eluent) (photos provided by the authors)
13. Product
1 is >99% pure as measured by
1H NMR
pdf using 1,3,5-trimethoxybenzene (Sigma Aldrich, 99%) as internal standard. mp = 87-89 ℃.
1H NMR
pdf (500 MHz, CDCl
3) δ 7.87 - 7.80 (m, 2H), 7.75 - 7.67 (m, 2H), 3.75 (t,
J = 6.8 Hz, 2H), 3.15 (t,
J = 7.2 Hz, 2H), 2.23 (p,
J = 7.0 Hz, 2H).
13C NMR
pdf (126 MHz, CDCl
3) δ 168.6, 134.5, 132.4, 123.7, 39.0, 33.0, 1.6. FTIR (KBr pellet): 2978, 2924, 1763, 1706, 1400, 1311, 1199, 1054, 723 (cm
-1). HRMS (ESI) m/z, calcd for [C
11H
11INO
2] ([M+H]
+): 315.9829, found: 315.9827 Anal. calcd for C
11H
10INO
2: C, 41.93; H, 3.20; I, 40.27; N, 4.45. Found: C, 41.66; H, 3.17; I, 40.00; N, 4.37.
pdf14. A second reaction on the same scale provided 6.01 g (95%) of product (
1) in >99% purity.
15.
2,9-dimethyl-4,7-diphenyl-1,10-phenanthroline (
bathocuproine) (98%) was purchased from Sigma Aldrich and used as received.
16.
Nickel(II) bromide 2-methoxyethyl ether complex was purchased from Sigma-Aldrich and used as received. Alternatively, Nickel(II) bromide ethylene glycol dimethyl ether complex, purchased from Sigma Aldrich (97%), can also be used.
17.
Nickel(II) bromide 2-methoxyethyl ether complex is hygroscopic and turns into a semi-solid upon standing under air at rt (22 ℃)..
18. Weighing paper (4" x 4") was purchased from Fisher Scientific.
19.
Dioxane (99.9%) was purchased from Fisher Scientific and used as received.
20. 1-Dram vial and the Teflon-coated polypropylene cap were purchased from Chemglass Life Sciences.
21.
Potassium tert-butoxide (98+%) was purchased from Sigma Aldrich and used as received.
22.
Potassium tert-butoxide is hygroscopic and turns into a semi-solid upon standing under air at RT.
23. 20-mL vials were purchased from Fisher Scientific and the DWK Life Sciences Wheaton Teflon-coated polypropylene caps were purchased via Fisher Scientific.
24. The stirring should continue until all the solids form a fine suspension. If a clump of solid forms, it can be broken up with a metallic spatula.
25.
Nitrocyclohexane (97%) was purchased from Neta (Spectrum Chemicals) and used as received.
26. A slight excess of nitroalkane (0.1 equiv) relative to the base provides higher yields.
27.
Tris[2-phenylpyridinato-C2,N]iridium(III) (Ir(ppy)
3) (99%) was purchased from Sigma-Aldrich and used as received. Alternatively, it can be synthesized by a published Organic Syntheses procedure.
228. Alternatively, the authors note
NiBr2·bathocuproine complex (
3) can be made as follows:
2,9-dimethyl-4,7-diphenyl-1,10-phenanthroline (
bathocuproine) (1.08 g, 3.0 mmol, 1.0 equiv),
nickel(II) bromide 2-methoxyethyl ether complex (
NiBr2·diglyme) (1.06 g, 3.0 mmol, 1.0 equiv), and
dioxane (20 mL) are added to a 50-mL round-bottom flask under ambient temperature (22 ℃) and stirred for 12 h. The mixture is diluted with
hexanes (20 mL), and the pink solids are filtered and washed sequentially with
hexanes (5 mL) and
diethyl ether (5 mL). The solids are dried under vacuum (0.05 mm Hg) for 24 h to provide
3 (1.73 g, 100%) which is used without further purification as the pre-catalyst in the alkylation reaction.
Figure 6. Top left: mixture of ligand and dioxane; Top middle: reaction turns pink after addition of the nickel source; Top right: the solids are filtered on a sintered funnel; Bottom: product 2 as a pink solid (Photos provided by authors)
29. The Penn PhD Photoreactor M2 was purchased from Sigma Aldrich. A second reaction on the same scale with this setup provided 1.12 g (70 %) of product (
2) in 98.1 % purity.
30. Alternatively, the authors also showed the reaction can be conducted under Kessil lamp for 24 h. The vial was placed on a stir plate with two 40 W blue LED (450 nm) lamps on each side (6 cm away from the vial) and a 4" desktop fan is placed on top (6 cm away from the vial) to dissipate the heat. A reaction on the same scale with this set up provided them 1.20 g, 76% yield of product (
2) >99% purity. A second reaction with this set up and scale provided them 1.16 g, 73% yield of product (
2) 98% purity.
Figure 7. Setup with Kessil lamps and a fan, light off and light on. (Photos provided by authors)
32.
Celite (545-mesh powder, non-acid washed) was purchased from Fisher Scientific and used as received.
33. The course of the chromatography can be monitored by TLC (EMD Millipore, TLC
silica gel 60 F
254). Product (
2): Rf = 0.35, 80:20
hexanes/EtOAc, visualized with UV lamp (254 nm).
2-(3-iodopropyl)isoindoline-1,3-dione (
1): Rf = 0.43, 80:20
hexanes/EtOAc, visualized with UV lamp (254 nm).
Figure 8. TLC of crude reaction and starting material (80:20 hexanes/EtOAc as eluent) (Photos provided by authors)
34. Product (
2) is >99% pure as measured by
1H NMR
pdf using 1,3,5-trimethoxybenzene (Sigma Aldrich, 99%) as internal standard. mp = 109-111 ℃.
1H NMR
pdf (500 MHz, CDCl
3) δ 7.85 - 7.74 (m, 2H), 7.68 (dd,
J = 5.5, 3.0 Hz, 2H), 3.61 (t,
J = 7.0 Hz, 2H), 2.40 - 2.31 (m, 2H), 1.85 - 1.78 (m, 2H), 1.70 - 1.43 (m, 7H), 1.34 (tdd,
J = 15.8, 7.5, 3.6 Hz, 2H), 1.23 (dddd,
J = 14.5, 12.5, 6.4, 4.2 Hz, 1H).
13C NMR
pdf (126 MHz, CDCl
3) δ 168.1, 133.9, 131.9, 123.2, 90.9, 37.6, 37.4, 33.9, 24.7, 22.6, 22.2. FTIR (KBr pellet): 2941, 2871, 1768, 1718, 1533, 1399, 1383, 1025, 722 (cm
-1). HRMS (ESI) m/z, calcd for [C
17H
21N
2O
4] ([M+H]
+): 317.1496. Found: 317.1493. Anal. calcd for C
17H
20N
2O
4: C, 64.54; H, 6.37; N, 8.86. Found: C, 64.79; H, 6.45; N, 8.87.
pdf35. The size and shape of the reaction vessel is important for efficient mixing, as the nitronate precipitates as a heterogenous salt, and adequate photon distribution. Simple scintillation vials have a large enough surface area to allow for maximum light exposure from commercial kessil lamps. As such, the reported procedure is limited to a 1.2 gram scale to fit within the scintillation vial and still stir effectually. A limited scale for maximum surface area to volume ratio is precedented in Organic Syntheses papers utilizing photochemistry.
3
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Nitroalkanes are highly versatile functional groups in organic synthesis and have a broad spectrum of uses and transformations.
4 The presence of an acidic α-proton (pKa ≈ 10 in H
2O) allows for the use of mild bases for deprotonation. The resulting nitronate anion can then be coupled with a diverse array of electrophiles through well-developed reactions, such as Henry reactions, Michael additions, and palladium-catalyzed arylations and allylations.
4,5 However,
C-alkylation of nitroalkanes with alkyl halides has been historically challenging due to the predominance of
O-alkylation with this class of electrophiles, which leads to the oxidation of the alkyl halide and reduction of the nitroalkane (Scheme 1, top).
6 Nitronate anions, however, undergo
C-alkylation with alkyl radicals at rates comparable to the addition of alkyl radicals to α,β-unsaturated carbonyls.
7 Taking advantage of this mode of reactivity, several radical precursors such as alkyl cobalt, alkyl mercury, and alkyl pyridinium reagents have been successfully coupled with nitroalkanes to furnish
C-alkylated products.
8 Nonetheless, the use of alkyl halides is more desirable given their numerous advantages over the aforementioned reagents; such as availability, atom-economy, and ease of handling and synthesis.
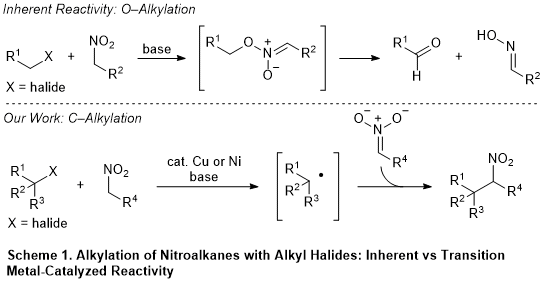
During the past few years, our group has investigated this radical-based approach by utilizing transition-metal catalysis. Alkyl radicals can be generated through low valent metal-mediated abstraction of halides from alkyl halides (Scheme 1, bottom). We discovered copper-catalyzed conditions for the alkylation of alkyl bromides bearing adjacent radical stabilizing groups (benzyl, carbonyl, and nitrile).
9 These conditions have expanded the scope of nitroalkane alkylations to a great extent and provided general routes for rapid synthesis of complex nitroalkanes. These copper-catalysted conditions, however, failed to provide meaningful yields with alkyl halides lacking the adjacent radical stabilizing groups, i.e.
unactivated alkyl halides. Our efforts toward finding a more general catalytic system led to the discovery of conditions using Ni/Et
2Zn that can efficiently couple unactivated alkyl iodides with nitronate anions.
10 Additionally, these conditions show excellent functional group tolerance, thus significantly broadened the scope for synthesis of complex nitroalkanes.
Although the scope of the Ni/Et2Zn catalytic conditions was remarkable with regard to the tolerance of a wide range of alkyl iodides (1°, 2°, and 3° alkyl iodides all are compatible with the alkylation reaction), the scope was limited to 1° nitroalkanes. Secondary nitroalkanes only afforded trace amounts of C-alkylated products, and nitromethane suffered from modest yields and significant overalkylation. Moreover, the use of air- and moisture-sensitive Et2Zn as the initial reductant rendered the bench-top setup of the reaction (i.e., those not requiring a glovebox) challenging, requiring a sophisticated setup that was less practical for the general chemistry community. Therefore, we sought to find catalytic conditions to address these issues.
We hypothesized that the presence of Lewis acidic zinc byproducts might have detrimental effects on the catalyst by potentially competing with nickel for the
bathocuproine ligand. We envisioned that a photoredox catalyst might act as the initial reductant after excitation by light, as proposed in the literature.
11 Due to their coordinatively stable structure, the photoredox catalysts would not compete for ligands, therefore increasing the reactivity of the system.
12 Indeed, this hypothesis proved fruitful when Et
2Zn was replaced with Ir(ppy)
3 and the reaction was irradiated with blue LED light, leading to the protocol outlined in this procedure. Secondary nitroalkanes and nitromethane, as well as primary nitroalkanes, can now be efficiently coupled with unactivated alkyl iodides. Interestingly, other classes of alkyl halides, such as benzyl halides, α-halocarbonyls, and α-halonitriles, are also competent electrophiles, which further demonstrates the generality of this reaction. More importantly, the current conditions offer significantly improved air and moisture stability compared to our previous copper- and nickel-catalyzed conditions, allowing the reaction to be set up without need for anerobic or anhydrous solvents.
13In the reported example here, the nickel pre-catalyst is formed in situ by simply mixing commercially available reagents, NiBr2·diglyme and bathocuproine. However, this complex can also be isolated and used as an air-stable and non-hygroscopic solid (see notes). The alkyl iodides can be synthesized in high yields from the corresponding alcohols using a variety of well-known reactions. In this case, we used the Appel reaction, which provided the primary alkyl iodide (1) in excellent yields. The resulting nickel pre-catalyst, and 1° and 2° alkyl iodides are stable indefinitely at -20 ℃. The Ni/Ir dual-catalyzed reaction of a typical 2° nitroalkane, nitrocyclohexane, with the primary alkyl iodide (1), affords the sterically congested 3° nitroalkane product (2) in good yields. We should also emphasize that all the reactions were set up on the bench under air with no caution for oxygen and moisture exclusion. The only precaution that is required, at least in our hands, is to ensure that the reactor is sealed prior to irradiation and that the headspace in the reactor is not large (we assume that under these conditions, ambient oxygen is consumed in a photoredox reaction, perhaps with the solvent, prior to the initiation of the alkylation reaction, but we have been unable to verify this hypothesis). This is further advantageous as only common glassware and equipment found in most organic and medicinal labs are sufficient.
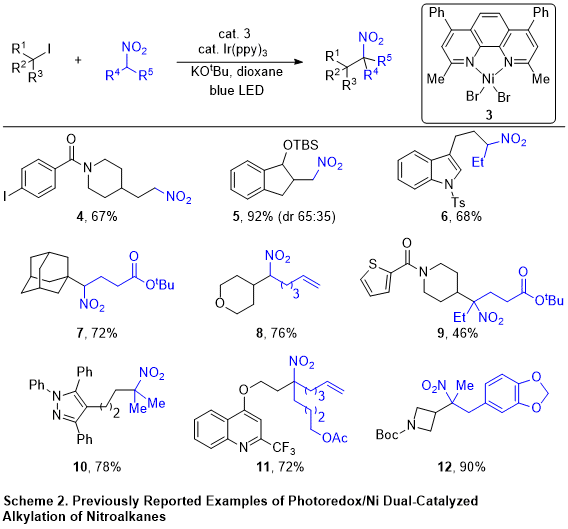
This method is general for coupling a broad spectrum of alkyl iodides and nitroalkanes. Nitromethane, 1° and 2° nitroalkanes, and 1°, 2°, and 3° alkyl iodides can be utilized (Scheme 2). The method also shows excellent functional group tolerance, including heterocycles. The nitro functionality of the alkylated products can be reduced with a variety of methods, offering access to sterically congested 2° and 3° amines.
13Importantly, only a slight excess of primary (1.1 equiv) or secondary (1.5 equiv) nitroalkane is required for good yields in this intermolecular reaction. This is particularly advantageous in the context of total synthesis and medicinal chemistry, as it allows the coupling of two complex fragments in near equimolar amounts. In the case of nitromethane, however, an excess amount (10-15 equiv.) is necessary to minimize the amount of the bis-alkylated product.
In conclusion, we have reported a general method for alkylation of nitroalkanes with alkyl halides. The broad scope and excellent functional group tolerance of this reaction enables the formation of complex nitroalkanes, which can be further converted to sterically encumbered and medicinally relevant alkyl amines. Finally, the robustness of the reaction allows it to be set up without need to for anerobic or anhydrous solvents, making it convenient and broadly useful to the chemistry community.
Appendix
Chemical Abstracts Nomenclature (Registry Number)
(2,9-dimethyl-4,7-diphenyl-1,10-phenanthroline) Nickel(II) Bromide: Nickel, dibromo(2,9-dimethyl-4,7-diphenyl-1,10-phenanthroline-ΚN1,ΚN10)-, (T-4)-; (326822-02-0)
2,9-dimethyl-4,7-diphenyl-1,10-phenanthroline: 1,10-phenanthroline, 2,9-dimethyl-4,7-diphenyl-; (4733-39-5)
Dioxane: 1,4-dioxane; (123-91-1)
Nickel(II) bromide 2-methoxyethyl ether complex: Nickel, dibromo[1,1'-(oxy-ΚO)bis[2-(methoxy-ΚO)ethane]]-; (312696-09-6)
Hexanes: Hexanes; (110-54-3)
Diethyl ether: Ethane, 1, 1'-oxybis-; (60-29-7)
2-(3-iodopropyl)isoindoline-1,3-dione: 1H-Isoindole-1,3(2H)-dione, 2-(3-iodopropyl)-; (1)(5457-29-4)
triphenylphosphine: Phosphine, triphenyl-; (603-35-0)
Imidazole: 1H-Imidazole; (288-32-4)
Dichloromethane: Methane, dichloro-; (75-09-2)
Iodine: Iodine; (7553-56-2)
2-(3-hydroxypropyl)isoindoline-1,3-dione: 1H-Isoindole-1,3(2H)-dione, 2-(3-hydroxypropyl)-; (883-44-3)
Anhydrous magnesium sulfate: Sulfuric acid magnesium salt; (7487-88-9)
Ethyl acetate: Acetic acid ethyl ester; (141-78-6)
Silica gel; (112926-00-8)
Potassium tert-butoxide: 2-Propanol, 2-methyl-, potassium salt; (865-47-4)
Nitrocyclohexane: Cyclohexane, nitro-; (1122-60-7)
Tris[2-phenylpyridinato-C2,N] iridium(III); Iridium, tris[2-(2-pyridinyl-ΚN)phenyl-ΚC]-, (OC-6-22)-; (94928-86-6)
Celite; (61790-53-2)

|
Sina Rezazadeh was born in Ramsar, a small town in north of Iran. He obtained his Pharm.D. in 2013 from Tehran University of Medical Sciences. In 2020, he received his PhD in Organic Chemistry from the University of Delaware under Prof. Donald Watson. His research focused on developing transition metal-catalyzed alkylations of nitroalkanes and amides. He currently works at Prelude Therapeutics in Wilmington, Delaware. |

|
Allyssa Conner was born and raised in Plymouth Meeting, Pennsylvania. She obtained her B.S. in Biochemistry at Lafayette College in 2018 and her PhD in Organic Chemistry from the University of Delaware. Her research focused on developing transition metal-catalyzed methods to synthesize and utilize silicon-containing compounds and nitroalkanes. She currently works at Ashland Inc. in Wilmington, Delaware. |

|
Donald Watson received his BS in Chemistry from UC San Diego and PhD in Organic Chemistry from UC Irvine. He then completed postdoctoral training at UC Berkeley and MIT. In 2009, he joined the faculty in the Department of Chemistry and Biochemistry at the University of Delaware, where he has risen to the rank of Professor. His research interests focus on organic and organometallic chemistry, as well as the discovery and understanding of new synthetic methods. |

|
Neetu Sharma is from North India, completed her Master of Science at the National Institute of Technology, Tiruchirappalli. Under the guidance of Dr. R. Karvembu and Dr. Seenuvasan Vedachalam, her research centered on developing metal-free cross-coupling. In 2022, she joined the Brown Lab, excited to expand her scientific journey into the fascinating world of photochemistry. |
Copyright © 1921-2025, Organic Syntheses, Inc. All Rights Reserved