Org. Synth. 2025, 102, 128-142
DOI: 10.15227/orgsyn.102.0128
C(sp2)-S Cross-Coupling Reactions with Adaptive Dynamic Homogeneous Catalysis: Synthesis of Ethyl 4-((3-Ethoxy-3-oxopropyl)thio)benzoate
Submitted by Florence Babawale
1, Indrajit Ghosh
1*, Burkhard König
1*
Checked by Nicholas D. D'Arcy-Evans, Evan Rutter, and Darren J. Dixon
1. Procedure (Note 1)
A 50-mL Schlenk tube (Note 2) is charged with 4CzIPN (15.8 mg, 0.02 mmol) (Note 3), NiBr2•glyme (154.3 mg, 0.5 mmol) (Note 4), ethyl 4-bromobenzoate (2.29 g, 10.0 mmol, 1.0 equiv.) (Note 5), and ethyl 3-mercaptopropionate (2.01 g, 15.0 mmol, 1.5 equiv) (Note 6) (Figure 1). A PTFE-coated magnetic stirring bar (25 × 10 mm, polygonal) is then added, and the vial is closed with a rubber septum. Dimethylacetamide (DMA, 20 mL) (Note 7) is then added to the vial using a plastic syringe and needle (diameter = 0.8 mm) through the rubber septum, giving a black suspension (Figure 1). The reaction mixture is then introduced to a nitrogen atmosphere using the following method: the sealed vial is placed under vacuum (~9 mm Hg) for approximately 30 seconds with a syringe needle (diameter = 0.60 mm) and backfilled with nitrogen for approximately 30 seconds (Note 8). This process is repeated three times. Parafilm is then wrapped around the rubber septum atop the Schlenk tube to ensure a seal.

Figure 1. Preparation of reaction mixture, stepwise addition of reagents: A. 50 mL Schlenk tube; B. Addition of 4CzIPN (15.8 mg); C. Addition of NiBr2•glyme (154.3 mg); D. Addition of ethyl 4-bromobenzoate (2.29 g); E. Addition of ethyl 3-mercaptopropionate (2.01 g); F. Addition of a stirring bar (25 × 10 mm, polygonal) and sealing of the reaction vial with a rubber septum; G. Reaction mixture under nitrogen with sealed parafilm containing stirring bar, reagents and solvent (DMA, 20 mL) (Photos are provided by Checkers)
The reaction mixture is then irradiated with one 40-Watt PR160L Kessil Lamp (λmax = 456 nm) (Note 9) at full intensity at a distance of approximately 3 cm with stirring at 600 rpm (Figure 2). A strong flow of nitrogen through an inverted cone directed at the flask was used to maintain a temperature of approximately 30 ℃ (Note 10).
Figure 2 (Checker photos). Photochemical reaction setup. A. Reaction vial at a distance of about 3 cm from the Kessil lamp; B. Reaction vial with inverted funnel channelling a strong flow of nitrogen as a source of cooling C. Color of the reaction mixture after completion (24 h). (Photos are provided by Checkers)
After 24 h (Note 11), stirring is stopped, the reaction vial is opened, and the contents (without the stirring bar) are transferred into a 250-mL separatory funnel. Deionized water (10 mL) and EtOAc (10 mL) (Note 12) are used to rinse the reaction vial to ensure a quantitative transfer. Water (40 mL) is then added to the separatory funnel (Figure 3). The aqueous layer is extracted with EtOAc (3 × 40 mL) (Figure 3B). The combined organic layer is washed with brine (20 mL) (Figure 3C-D). Afterward, EtOAc (5 mL) is used to rinse the walls of the separatory funnel using a glass pipette. The combined organic layer is dried over 7 g of anhydrous magnesium sulfate (Note 13) for 15 min. After drying, the organic layer is filtered through a powder funnel with a fluted filter paper (Note 14). The magnesium sulfate is rinsed with EtOAc (3 × 10 mL), the combined organic layer is collected, and TLC of the reaction mixture is performed (Note 15). The organic layer is then concentrated on a rotary evaporator (40 ℃, 240 to 20 mm Hg) to give the crude material, a yellow-orange oil.

Figure 3. Picture of the extraction process. A. Separated organic and aqueous layers after addition of water (40 mL) for the first extraction; B. Separated organic and aqueous layers during the last extraction; C. Separated organic and aqueous layers during the brine wash; D. Combined organic layers; (E and F) The TLC plate was eluted with 15% (v/v) ethyl acetate/hexane and visualized using 254 nm UV light. The TLC was then stained with potassium permanganate to visualize the by-product of the reaction, i.e. diethyl 3,3'-disulfanediyldipropionate (Rf = 0.38) (Note 15). Lane A contains a standard of ethyl 4-bromobenzoate (Note 5), Rf = 0.48. Lane B is a mixture of ethyl 4-bromobenzoate and the reaction mixture. Lane C contains the reaction mixture, showing mainly the desired product, Rf = 0.25 (Photos are provided by Checkers)
For the purification step, column chromatography is carried out. A column with a length of 27 cm with a 6 cm diameter is used. The column is dry packed in the following manner: first with cotton wool, sea sand (20 g), silica gel (163 g) (Note 16), and again sea sand (40 g) to protect the surface of the silica (Figure 4A). The surface is leveled horizontally at each stage. The column is equilibrated using 540 mL solution of 2.5% (v/v) ethyl acetate/hexane (Note 17). The crude material is transferred onto the compacted column with a glass pipette. The flask is rinsed with 2.5% (v/v) ethyl acetate/hexane (3 × 1 mL), and the washings are added to the column. The column is eluted with a slow gradient starting using 2.5% (v/v) ethyl acetate/hexane with an increment of 2.5% to 5% until 20% ethyl acetate/hexane (Figure 4B). A total of 2.1 L of solvent is used (Note 18). The tubes (Note 19) containing pure desired product are collected in a 1-L round-bottomed flask and concentrated on a rotary evaporator (40 ℃, 330 to 20 mm Hg). The pure product is then transferred into a 100-mL round-bottomed flask. The 1-L flask is rinsed with EtOAc (2 × 5 mL) into the 100-mL flask and then re-concentrated on a rotary evaporator (40 ℃, 240 to 20 mm Hg). The pure product is then transferred into a 25 mL screw-cap vial. The 100-mL flask is then rinsed with CH2Cl2 (2 × 1 mL) into the vial, which is then concentrated on the rotary evaporator (40 ℃, 375 to 20 mm Hg).
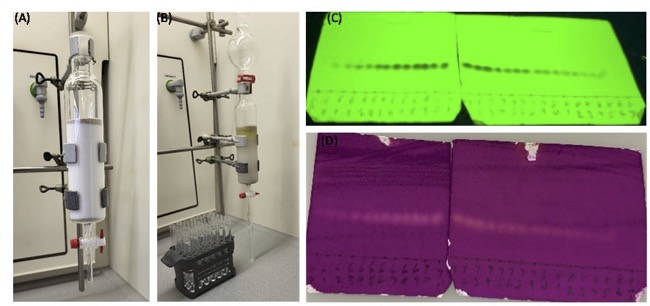
Figure 4 Pictures of column chromatography. A. The packing of the column for flash column chromatography; B. Purification of the crude material; C. and D. TLC analysis of the column fractions (Note 19). (Photos are provided by Authors)
The resulting colorless oil is kept under a high vacuum (25 ℃, ca. 4 mmH g) for 4 h to afford pure product ethyl 4-((3-ethoxy-3-oxopropyl) thio)benzoate (2.44 g, 86% yield) (Notes 20 and 21) with 97% purity determined by qNMR analysis (Notes 22 and 23). A second run provided (2.60 g, 92% yield) with 97% purity determined by qNMR analysis.
Figure 5. Ethyl 4-((3-ethoxy-3-oxopropyl)thio)benzoate
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/about/governance/committees/chemical-safety.html assessment.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
ethyl 3-mercaptopropionate,
ethyl 4-bromobenzoate,
nickel(II) bromide ethylene glycol dimethyl ether complex,
dimethylacetamide,
dimethyl fumarate,
ethyl acetate,
hexane,
MgSO4, silica gel. Additionally, proper safety glasses, such as orange filter safety glasses, should be worn, and individuals should be aware of the hazards associated with the use of a 40
-Watt Kessil Lamp.
2. The Schlenk tube (B14/23, clear glass, 50 mL, 110 mm × 30 mm, with PTFE needle-valve stopcock) was purchased from Fischer Scientific and was used as received. The side arm was sealed during the reaction to mimic the microwave vial used by the submitters.
3. 4CzIPN (97%) was purchased from BLDpharm and used as received.
4.
Nickel(II) bromide ethylene glycol dimethyl ether complex (98%) was purchased from BLDpharm and used as received.
5.
Ethyl 4-bromobenzoate (98%) was purchased from Fluorochem and used as received. It is important to note that depending on the supplier, trace amounts of impurities may influence the reaction outcome, for example, slow reaction kinetics. In such cases, distillation of the electrophile is recommended.
6.
Ethyl 3-mercaptopropionate (99.1%) was purchased from BLDpharm and used as received.
7.
N,N-Dimethylacetamide, anhydrous (99.8%) was purchased from Thermo Fischer Scientific and used as received.
8. Degassing of the reacti
on mixture was carried out with stirring at 200 rpm.
10. The authors report that "a slight increase in the reaction temperature has no detrimental effect on the reported C(sp
2)-S cross-coupling reaction outcome and generally increases the reaction kinetics rate."
11. The reaction progress can be monitored either by using GC and GC-MS analysis (in this case, substrate consumption and the formation of the desired product) or by using TLC of the crude reaction mixture (in this case, mainly substrate consumption) eluting with 15% (v/v)
EtOAc/
hexane and visualized with 254 nm UV light. The TLC plate was stained using
potassium permanganate to visualize the side product of the reaction, diethyl 3,3'-disulfanediyldipropionate. R
f (side product) = 0.38. R
f (product) = 0.25. After 3 h, evidence of starting material was still observed. After 8 h, the expected brown to cyan color change was observed, but the starting material still remained. Only after 24 h was full consumption of starting material observed. It is to be noted here that the reaction time depends on the purity of the starting materials (in this case,
ethyl 4-bromobenzoate as an electrophile). See note 5 for further details.
12.
Ethyl acetate (99.7 %) was purchased from Sigma Aldrich and used as received.
13. Anhydrous
MgSO4 (98%) was purchased from Thermo Fisher Scientific and used as received.
14. Filter paper was purchased from Fisher Scientific (QL100, diameter
=110 mm). Powder funnel, diameter
=80 mm.
15. The TLC plate was eluted with 15% (v/v)
ethyl acetate/
hexane and visualized using 254 nm UV light. Subsequently, the TLC was stained with
potassium permanganate to visualize the side product of the reaction, diethyl 3,3'-disulfanediyldipropionate (R
f = 0.38). It is important to note that depending on the (het)aryl halides, the product, and the disulfide may appear very close to each other, thus a quick staining with
potassium permanganate is recommended.
16. Silica gel (60 Å, 230 - 400 mesh) was purchased from Sigma Aldrich and used as received.
17.
Hexane (≥ 97 %) was purchased from Sigma-Aldrich and used as received.
18. 2.5% (v/v)
ethyl acetate/
hexane (300 mL), 5% (v/v)
ethyl acetate/
hexane (300 mL), 7.5% (v/v)
ethyl acetate/
hexane (300 mL), 10% (v/v)
ethyl acetate/
hexane (300 mL), 15% (v/v)
ethyl acetate/
hexane (600 mL) and 20% (v/v)
ethyl acetate/
hexane (300 mL).
19. The eluent was collected into 16 x 160 mm test tubes (approximately, 20 mL per fraction). Fraction collection began at 10% (v/v)
ethyl acetate/
hexane. Fractions containing the product (fractions 65 - 84) were identified by TLC analysis using 15% (v/v)
ethyl acetate/
hexane. Each test tube containing pure product was washed once with
EtOAc (1 mL) to ensure quantitative transfer. Impure fractions containing both the product and the side product diethyl 3,3'-disulfanediyldipropionate (fractions 62 - 64) were discarded.
20. All NMR analysis was performed using chloroform-D (99.8%) as the solvent, which was purchased from Sigma-Aldrich and used as received.
21. Characterization data of product
1:
1H NMR
pdf (CDCl
3, 400 MHz): 7.94 (2H, d,
J = 8.7 Hz), 7.32 (2H, d,
J = 8.8 Hz), 4.36 (2H, q,
J = 7.1 Hz), 4.15 (2H, q,
J = 7.1 Hz), 3.24 (2H, t,
J = 7.4 Hz), 2.66 (2H, t,
J = 7.4 Hz), 1.38 (3H, t,
J = 7.1 Hz), 1.25 (3H, t,
J = 7.1 Hz).
13C NMR
pdf (CDCl
3, 100 MHz): 171.6, 166.3, 142.6, 130.2, 127.8, 127.3, 61.1, 60.9, 34.1, 27.5, 14.5, 14.3. HRMS: calculated for [M + H]
+ C
14H
19O
4S 283.0999; found 283.1001. Main signals of IR (neat, cm
-1) 2982, 1714, 1594, 1400, 1370, 1273, 1244.
22. The purity of the product was determined by
qNMR
pdf analysis using 34.2 mg of
ethyl 4-((3-ethoxy-3-oxopropyl)thio)benzoate and 16.6 mg of
dimethyl fumarate as an internal standard in CDCl
3. The purity was calculated as 97%.
23.
Dimethyl fumarate (99%) was purchased from Thermo Fisher and used as received.
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Cross-coupling reactions play a central role in modern organic chemistry, facilitating the formation of chemical bonds that are essential for the synthesis of a wide range of molecules.
2,3 From simple intermediates to complex biomolecules, these reactions allow the efficient construction of molecular architectures that are crucial in various fields ranging from pharmaceuticals to materials science. Although the range of reported (het)aryl halides and nucleophile coupling partners is very large, with different transition metals (e.g. Pd, Ni, Cu, and others) and different protocols, the reaction conditions vary considerably between classes of compounds, requiring renewed case-by-case optimisation of the reaction conditions.
4 Our recent work on adaptive dynamic homogeneous catalysis (AD-HoC) presents a groundbreaking approach that enables the formation of diverse chemical bonds under highly predictable reaction conditions.
5 This is synthetically demonstrated in nine different bond-forming reactions (in this case C(sp
2)-S, Se, N, P, B, O, C(sp
3, sp
2, sp), Si, Cl) covering hundreds of synthetic examples. This advance holds great promise for streamlining synthetic routes and thus accelerating the development of drugs, medicines and functional materials.
Among various cross-coupling reactions, C(sp
2)-S cross-coupling reactions have recently emerged as crucial transformations leading to the production of valuable products, including pharmaceuticals and materials.
6,7 While other photoredox-catalysed synthetic methods have been reported that allow the formation of the C(sp
2)-S bond, they are generally limited to the use of aryl iodides
8 and require the presence of various ligands, transition metal photocatalysts and additives such as pyridine to improve the synthetic method
9. Photocatalyst and nickel-free methods are also known for C(sp
2)-S bond-forming reactions via EDA complexes in the presence of a base, but are generally limited to the use of thiophenols as coupling partners.
10 Our method allows the use of all different types of thiols as nucleophiles for C-S bond-forming reactions under traditional ligand, base, and additional additive-free reaction conditions. The simplicity of the reaction conditions and the general tolerability of the reactions allow easy synthesis of various drug intermediates and late-stage functionalization of various biomolecules, including the synthesis of cefapirin precursor or S-arylation of glutathione or tiopronin, among others.
5 The cross-coupling reaction conditions are generally effective for a range of (het)aryl bromides and tolerate a variety of functional groups for such transformations (see Figure 6), with the exception of certain functional groups such as free amines. In terms of steric hindrance, the reactions are generally well tolerated in the electrophile, but highly ortho-substituted electrophiles, e.g. di-ortho-substituted electrophiles, are generally less effective for such transformations. The steric hindrance in the nucleophile is generally less problematic compared to the range of electrophiles. For example, 2,6-dimethylbenzenethiol can be easily coupled to (het)aryl bromides to give the desired product in good to excellent yields.
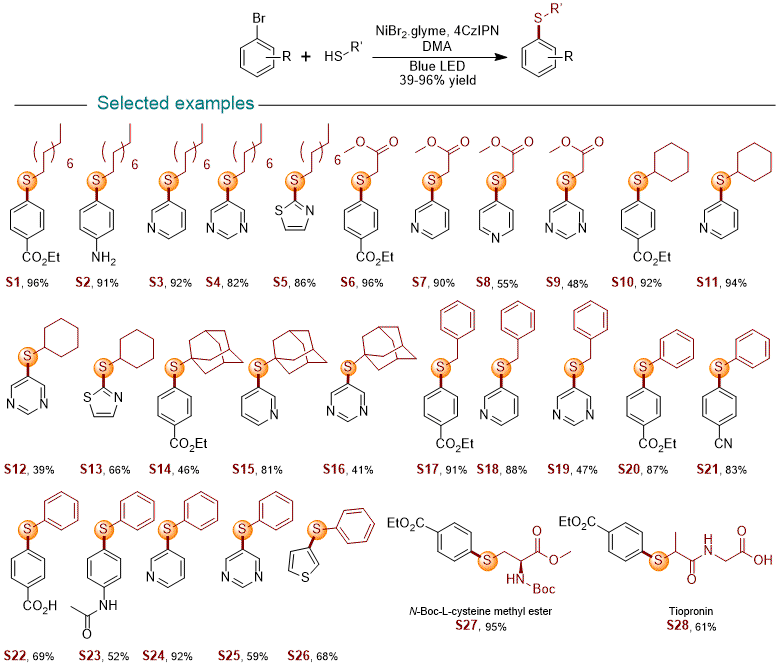
Figure 6. Selected substrate scope for photoredox catalytic C(sp2)-S bond formation reactions
It is to be noted here that we have previously reported the photochemical reaction in an in-house photochemical reaction setup.
5 Recognising that different research groups use different photochemical setups, for the sake of generality we have used Kessel lamps for the synthesis of the desired products. It is noteworthy that it has recently been shown that the yield and kinetics of the desired product are highly dependent on the use of different photochemical setups,
11 and reproducibility is becoming a major topic of discussion.
12 The simplicity of the reaction conditions allows for easy translation of the photochemical reaction to be carried out in different photochemical setups.
The reactions generally follow the Ni(I)/Ni(III) catalytic cycle for transformations. A number of different species are expected to be formed, which are in equilibrium. Under photocatalytic reaction conditions, one of the species is reduced to Ni(I), which can undergo oxidative addition with the aryl halides.
We believe that the simplicity of the reaction conditions, together with the possibility of easy gram-scale synthesis, will allow the use of this method in various laboratories and in pharmaceutical and other industries.
Appendix
Chemical Abstracts Nomenclature (Registry Number)
Ethyl 3-mercaptopropionate; (5466-06-8)
Ethyl 4-bromobenzoate; (5798-75-4)
Nickel(II) Bromide Ethylene Glycol Dimethyl Ether Complex; (28923-39-9)
Dimethylacetamide; extra dry with molecular sieves; (127-19-5)
Dimethyl fumarate; (624-49-7)

|
Florence Babawale obtained her B.Sc. from the University of Ghana in 2017 under the supervision of Dr. Richard B. Owoare. In 2021, she completed her M.Sc. degree from the Elite Network of Bavaria's Synthesis and Catalysis Master Programme under the guidance of Prof. Dr. Burkhard König at the University of Regensburg, Germany. Presently, she is pursuing her Ph.D. at the same institution. Her research focuses on utilizing photoredox catalysis for cross-coupling reactions, exploring both hydrogen atom transfer and nickel dual photoredox catalysis methodologies. |

|
Indrajit Ghosh received his Ph.D. in Chemistry in 2013 under the guidance of Prof. Werner M. Nau at Jacobs University Bremen, Germany. His postdoctoral research with Prof. Burkhard König at the University of Regensburg and Markus Antonietti at the Max Planck Institute of Colloids and Interfaces pioneered many innovative concepts in visible light photoredox catalysis. These include ConPET, SenI-ET, Chromo-selective, and AD-HoC catalysis, enabling the effective synthesis of value-added chemicals using visible light. |

|
Burkhard König received his Ph.D. from the University of Hamburg and continued his scientific education as a post-doctoral fellow with Prof. M. A. Bennett, Research School of Chemistry, Australian National University, Canberra, and Prof. B. M. Trost, Stanford University. Since 2000, he has been a full professor of organic chemistry at the University of Regensburg. His current research interests revolve around the use of visible light in organic synthesis. |

|
Evan Rutter is completing his MChem degree at the University of Oxford. He is currently working under the supervision of Prof. Darren Dixon for his Masters project in the field of Bifunctional Iminophosphorane (BIMP) catalysis for asymmetric synthesis. |

|
Nicholas D. D'Arcy-Evans was born in Johannesburg, South Africa in 1999. He completed his MChem at the University of St Andrews in 2023 under the supervision of Prof. Allan J. B. Watson, investigating photocatalytic methods for C-C bond formation. He is currently studying a DPhil in the group of Prof. Darren Dixon, focussing on the development novel strategies for molecular editing. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved