Org. Synth. 2025, 102, 143-155
DOI: 10.15227/orgsyn.102.0143
Preparation of (Z)-N-Phenoxybenzimidoyl Chloride
Submitted by Joseph P. Lokant, Ross M. Dare, and David A. Nagib*
1Checked by Souvik Adak and M. Kevin Brown
1. Procedure (Note 1)
A. N-phenoxybenzamide (1). A flame-dried, 1-L single-necked (24/40) round-bottom flask equipped with a 4.2 cm egg-shaped Teflon-coated magnetic stirrer is charged with benzohydroxamic acid (6.86 g, 50 mmol, 1.0 equiv, Note 2) and 500 mL of anhydrous ethanol (Note 3). The flask is covered with a rubber septum and the mixture is stirred (Note 4) for 1 min at room temperature (22-25 ℃, Note 5) to achieve a homogenous solution. Potassium tert-butoxide (6.17 g, 55 mmol, 1.1 equiv, Note 6) is added at once and the resulting mixture is stirred under a continuous flow of N2 (through an 18 G needle) for 1 h to obtain a clear colorless solution. Diphenyliodonium chloride (15.8 g, 50 mmol, 1.0 equiv, Note 7) is then added as a solid in approximately equivalent portions over 5 min using a spatula and the reaction is kept stirring under N2 for 16 h at room temperature (22-25 ℃). During this time, the reaction mixture turns into a turbid pale brown solution (Figure 1).

Figure 1. A. After the addition of KOtBu. B. After the addition of Ph2ICl. C. Reaction after 16 h
At this time, stirring is stopped and the contents of the vessel are allowed to settle prior to vacuum filtration through a 150-mL Buchner funnel with filter paper (7 cm, medium-fine porosity). The undissolved solids are washed with 50 mL of ethyl acetate and the combined filtrates are concentrated (20 mm Hg, 40 ℃, Note 8) to an oil. The residue is dissolved in 150 mL of ethyl acetate and 150 mL of deionized water and transferred to a 500-mL separatory funnel. After mixing, the aqueous layer is separated, and the organic fraction is washed with another 150 mL of deionized water. The aqueous layers are combined and extracted with 50 mL of ethyl acetate and then discarded. The resulting organic layers undergo vacuum filtration (0.01 atm) through a 150-mL Buchner funnel with filter paper (7 cm, medium-fine porosity) to remove any remaining undissolved solids. Following filtration, the organic layer is washed with 1 M sodium hydroxide (4 x 50 mL) and brine (saturated sodium chloride, 50 mL). These combined aqueous layers are transferred to a 1-L single-necked (24/40) round-bottom flask equipped with a 4.2 cm egg-shaped Teflon-coated magnetic stirrer. The solution is cooled to 0 ℃ (ice bath) before adding 150 mL of 1 M hydrochloric acid dropwise (over 10 min) via an addition funnel while stirring rapidly (Note 9). The precipitated fine solids are collected by vacuum filtration (0.01 atm) through a 150-mL Buchner funnel with filter paper (7 cm, medium-fine porosity) and then transferred to a 100-mL single-necked (24/40) round-bottom flask and dried under vacuum (<1 mm Hg) for 4 h to remove residual water. At this time, the crude isolated material (4.40 g) is taken forward in procedure B without further purification (Figure 2, Note 10).

Figure 2. A. Crude 1, B. Recrystallized 1
B. (Z)-N-phenoxybenzimidoyl chloride (2). A flame-dried 100-mL single-necked (24/40) round-bottom flask equipped with a 2.6 cm egg-shaped Teflon-coated magnetic stirrer is charged with phosphorus pentachloride (4.73 g, 22.7 mmol, 1.1 equiv, Note 11) and covered with a rubber septum. The headspace of the flask is evacuated and backfilled with N2 three times using an 18-G needle, and the vessel is cooled to 0 ℃ (ice bath). At this temperature, 41.2 mL (0.5 M with respect to 1) of dry chloroform (Note 12) is added and the solution is stirred for 5 min under a flow of N2. The crude N-phenoxybenzamide (4.39 g, 20.6 mmol, 1.0 equiv) is added quickly as a solid and the vessel is again placed under an inert atmosphere (Note 13). The ice bath is removed, and the reaction is allowed to stir for 45 min while gradually rising to room temperature (22-25 ℃). At this time, reaction completion is confirmed by thin-layer chromatography (Note 14). The flask is cooled to 0 ℃ (ice bath) and the reaction is quenched with 25 mL of deionized water and allowed to stir for 5 min. The contents are transferred to a 125-mL separatory funnel. The organic and aqueous layers are separated, and the aqueous layer is extracted with dichloromethane (3 x 25 mL). The combined organic layers are dried over anhydrous magnesium sulfate prior to vacuum filtration (0.01 atm) through a 150-mL Buchner funnel with filter paper (7 cm, medium-fine porosity) and concentrated by rotary evaporation (20 mm Hg, 25 ℃). The crude oil was purified by flash column chromatography using 100% hexanes as the eluent to afford 3.42 g (30% yield) of compound 2 as a colorless oil, which became a white solid after storing in the freezer overnight (99% purity, Note 15 and 16).

Figure 3. A. Reaction setup. B. Purified Compound 2
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/about/governance/committees/chemical-safety.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
benzohydroxamic acid,
ethanol,
potassium tert-butoxide,
diphenyliodonium chloride,
ethyl acetate,
sodium hydroxide,
hydrochloric acid,
phosphorus pentachloride,
chloroform,
dichloromethane,
magnesium sulfate, silica gel, and hexanes.
2.
Benzohydroxamic acid (98%) was purchased from Thermo Scientific and used as received.
3. Absolute
ethanol (200 proof) was purchased from Decon Laboratories, Inc. and used as received.
4. Stirring throughout these procedures is kept at a constant 850 rpm using an Ika RCT basic hotplate.
5. The term "room temperature" used throughout this manuscript refers to the range of temperatures between 22 ℃ and 25 ℃.
6.
Potassium tert-butoxide (98%) was purchased from Thermo Scientific and used as received. Upon initial addition of
potassium tert-butoxide, the solution will be cloudy and white. Within 5 min of stirring at room temperature, the solution will return to a homogeneous state.
7.
Diphenyliodonium chloride (98%) was purchased from Thermo Scientific and used as received. The rubber septum was only opened during the addition and otherwise kept closed under
N2 atmosphere. Upon addition to the reaction vessel, a color change from clear to yellow and finally to pale brown is observed. The solution will not return to a fully homogenous state, but the stirring will remain consistent and unaffected.
8. Rotary evaporation throughout these procedures was carried out using a Buchi R-100 Rotovap equipped with a Buchi B-100 heating bath and a Fischer Scientific Isotemp chiller.
9. Dropwise addition of the 1 M
hydrochloric acid was performed over 10 min. It is critical for this to be a slow addition for the desired precipitation of a fine solid that minimizes trapped impurities.
10. If desired, the crude solids can readily be purified to >97% before use in procedure B. In this case, the solids are transferred to a 125-mL Erlenmeyer flask and cooled to 0 ℃ (ice bath). Ice-cold diethyl ether (15 mL) is added, and the solids are gently triturated for 1 min using a spatula. The solvent, which will take a dark orange deep red color, is filtered away. The flask is rinsed with 20 mL of hexanes, which is also filtered to collect any remaining solids. The solids are transferred to a 100 mL single-necked (24/40) round-bottom flask containing a 2.6 cm egg-shaped Teflon-coated magnetic stirrer charged with 12 mL of
ethanol. The vessel is heated to 70 ℃ (aluminum block) while stirring rapidly. Once homogenous, 6 mL of deionized water is added dropwise over 3 min. Crystal formation will become visible and stirring is continued while the flask is removed from heat and allowed to cool to room temperature (20 min). The recrystallized solids are collected by vacuum filtration using a 150-mL Buchner funnel with filter paper (7 cm, medium-fine porosity). The flask is rinsed with 20 mL of hexanes, which is also filtered to collect any remaining solids. The solids are dried under vacuum (<1 mm Hg) for 2 h to provide 3.87 g of product
1 (>97% purity by quantitative
1H NMR using
ethylene carbonate as internal standard).
11.
Phosphorus (V) chloride (98%) was purchased from Alfa-Aesar. Prior to use in the reaction, the solid was transferred to a two-necked 250 mL round-bottom flask (24/40) and covered with rubber septa. In a fume hood, a stream of
N2 was blown over the solids through an 18-G needle (another 18-G needle was used as an outlet) for 24 h.
12. Extra dry
chloroform (99.9%) stabilized by amylene (<150 ppm) was purchased from Thermo Scientific and used as received. It is important to avoid the use of wet
chloroform or
chloroform stabilized by
ethanol, as the
phosphorus (V) pentachloride is hydrolyzed by water into
POCl3 and
HCl and by
ethanol into
POCl3,
HCl, and
EtCl.
13. Caution: Upon the addition of
N-phenoxybenzamide,
HCl extrusion occurs. The flask needs to be kept at 0 ℃ until bubble formation entirely ceases.
14. Thin-layer chromatography was performed using glass-backed silica plates with an eluent of 1:4
ethyl acetate:hexanes. The desired product has an Rf of 0.84 in this solvent mixture, whereas
N-phenoxybenzamide has an Rf of 0.39.
Figure 4. TLC of reaction B
15. Flash column chromatography was carried out using a 1.5 in ID x 12 in column prepared with 5 in of normal-phase silica gel. The crude product was dry-loaded onto ~ 5g of celite prior and 1 L of hexanes was used as the eluent. Purity of the isolated material was assessed by weight percent via quantitative
1H NMR using
ethylene carbonate as internal standard. A second run by the checkers provided similar results.
pdf16. The final product was isolated as a clear colorless oil which solidified to a white solid upon storage at -20 ℃, and the compound had the following characteristics:
1H NMR
pdf (500 MHz, CDCl
3) δ: 8.00 - 7.98 (m, 2H), 7.52 - 7.44 (m, 3H), 7.39 - 7.32 (m, 4H), 7.11 (tt,
J = 7.1, 1.4 Hz, 1H).
13C NMR
pdf (126 MHz, CDCl
3) δ: 159.0, 141.6, 132.4, 131.3, 129.6, 128.7, 127.7, 123.4, 115.1. IR (neat): 3062, 1592, 1486, 1263, 1205, 972, 946, 751, 686 cm
-1; HRMS (APCI) m/z calc'd for C
13H
11NO
35Cl [M + H]
+: 232.0524. Found: 232.0517.
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Oximes have emerged as privileged motifs in organic chemistry, as the ability to modulate their syntheses enables unique and versatile reactivity.
2 Among the multitude of valuable oxime scaffolds are imidoyl chlorides such as product
2.
3 This synthon provides rapid access to oxime imidates upon the addition of an
in situ-generated alkoxide nucleophile (Scheme 1). Early works of Taylor
4 and Ingold
5 examined the thermal decomposition of the
N-phenoxy imidates of phenols, wherein it was proposed that an imidyl radical intermediate is formed via homolysis of the N-O bond.
Scheme 1. Product 2 reacts with alkoxide nucleophiles to provide N-phenoxy imidates. Homolysis of the N-O bond yields imidyl and phenoxy radical intermediates
We have demonstrated the ability to generate this
N-centered radical under mild, photocatalytic conditions to enable selective functionalization of alcohols following conversion to their corresponding imidates. In the case of allyl imidates, the imidyl radical undergoes favorable 5-
exo-trig cyclization to yield an oxazoline heterocycle and an adjacent
C-centered radical. By introducing electrophilic radical traps, hydroamination, amino-alkylation, or amino-arylation products were realized (Scheme 2A).
6 Aiming to expand the scope of amino-functionalization products, we developed a dual-catalytic strategy wherein a copper catalyst traps the alkyl radical and enables cross-coupling with a nucleophile by reductive elimination (Scheme 2B).
7 With the goal of building further molecular complexity, we used a cobalt catalyst to invoke a metal-mediated hydrogen atom transfer (MHAT) of a significantly weakened C-H bond adjacent to the
C-radical - affording a vinyl oxazoline (Scheme 2C).
8 For aliphatic imidates, we observe a regioselective 1,5-hydrogen atom transfer (HAT), delivering an alkyl radical intermediate at the β-position relative to the pendant alcohol. With use of a copper catalyst bound with a chiral ligand, stereoselective reductive elimination provides access to aminated products of high enantioselectivity (Scheme 2D).
9
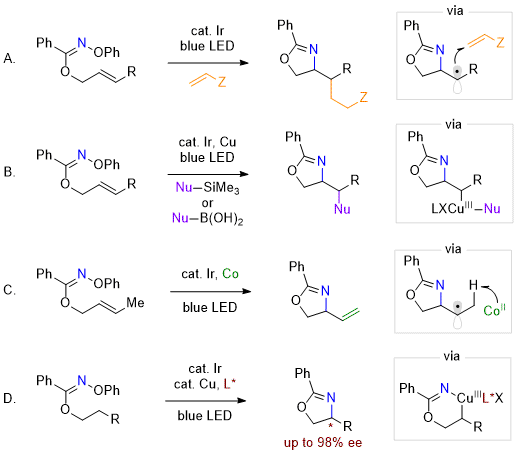
Scheme 2. (A) Amino-functionalizations of allyl imidates using electrophilic radical traps. (B) Amino-functionalizations of allyl imidates with nucleophilic cross-coupling partners. (C) Aza-Heck cyclization of allyl imidates to afford vinyl oxazolines. (D) Regio- and enantioselective C-H amination of alcohols
Whether utilizing this radical chaperone to afford diverse classes of 1,2-amino alcohols or to explore new catalytic reactivity, it is important that its synthesis be highly accessible and user-friendly. Rapid and reproducible production of N-phenoxybenzamide (1) is vital to achieving this goal. Two strategies exist for its synthesis: (1) a one-step arylation of benzohydroxamic acid with a diphenyliodonium salt, or (2) a three-step protocol from N-hydroxyphthalimide, involving a Chan-Lam coupling with phenylboronic acid, subsequent deprotection to the free amine, and then benzoylation (Scheme 3). Given the potential to greatly streamline access to this valuable compound, the one-step arylation was chosen as the ideal synthetic route.
Scheme 3. Viable retrosynthetic routes to N-phenoxybenzamide (1)
This arylation strategy has been utilized by Taylor
4 and Ingold.
5 In their syntheses, benzohydroxamate salts were pre-made and subjected to the arylation with diphenyliodonium in
tert-butanol at elevated temperatures. In employing this method, we observed that a significant number of inorganic impurities are carried over from the arylation step, which led to undesired isomerization of the imidoyl chloride and the subsequent oxime imidates. Additionally, further purification of the crude solid was time-consuming and non-trivial (multiple recrystallizations with significant material loss). Thus, we sought to develop milder conditions for this arylation. By switching to ethanol as the reaction solvent and using ambient room temperature instead of heating to reflux, we noticed a significant improvement in the quality of crude material obtained. Additionally, by using potassium tert-butoxide as base, an
in-situ deprotonation of benzohydroxamic acid was achieved, precluding an extra step in the protocol and affording a cleaner crude product
1 by slower, more selective reactivity.
The deoxy-chlorination of N-phenoxybenzamide (1) using phosphorus pentachloride (PCl5) to afford product 2 has also been dramatically improved. Traditionally, this transformation is performed by mixing the amide with excess PCl5 in carbon tetrachloride or chloroform for a minimum of six hours. Since PCl5 is highly reactive towards water and alcohol solvents, we hypothesized anhydrous chloroform (containing amylene, instead of ethanol, as stabilizer) would lead to greater reaction efficiency and reproducibility. To our delight, this change enabled lowering the amount of PCl5 to just 1.1 equivalents and decreased the reaction time to 45 minutes with complete conversion of 1 to yield product 2. Following flash column chromatography on silica gel, the imidoyl chloride is obtained as a white solid and importantly, with no isomerization observed.
In summary, this revised procedure enables reliable access to (Z)-N-phenoxybenzimidoyl chloride (2) in only 24 h from commercially available reagents. The conditions for the O-arylation of benzohydroxamic acid have been improved to give product 1 of high purity, precluding the need for further purification. Additionally, the deoxy-chlorination of this amide by PCl5 has been optimized to a 45-minute protocol, providing product 2 in high yield and with excellent purity.
Appendix
Chemical Abstracts Nomenclature (Registry Number)
Benzohydroxamic acid; (495-18-1)
Potassium tert-butoxide; (865-47-4)
Diphenyliodonium chloride; (1483-72-3)
Phosphorus pentachloride; (10026-13-8)

|
Joseph Lokant is from Oak Hill, West Virginia and received his B.S. in Biochemistry from West Virginia University in 2020. Joseph is currently a Ph. D. Candidate in the Nagib Lab at The Ohio State University, where his research focuses on the development of new regio- and enantioselective radical C-H functionalization reactions. |

|
Ross Dare is from Monroeville, New Jersey and attended Rowan University, graduating with a B.S. in Chemistry in 2018. Ross obtained his Ph. D. from The Ohio State University studying remote C-H functionalization of amines for desaturation and amination reactions. |

|
David Nagib grew up near Philadelphia, Pennsylvania as the eldest of four siblings in an Egyptian family with a strong love for teaching. His training is in asymmetric catalysis (with Scott Miller, Boston College, BSc 2006), photoredox organocatalysis (with Dave MacMillan, Princeton University, PhD 2011), and organometallic mechanisms (with Dean Toste, UC Berkeley, NIH Postdoc). Since 2014, David has taught at The Ohio State University, where he is the inaugural Harold and Betty Miller Professor of Organic Chemistry, as well as the College of Arts and Sciences Distinguished Professor of Chemistry and Biochemistry. His team's research on radical- and carbene- mediated C-H and C-O functionalization is dedicated to streamlining the synthesis of complex, medicinally relevant molecules. |

|
Souvik Adak was born in Tamluk, West Bengal, India. He pursued his bachelors study in chemistry (B.Sc. Hons) in Midnapore College (Autonomous). Then he moved to IIT Bombay to pursue his M.Sc. study in chemistry. During this time, he worked in Rh(III) catalyzed 'C-H' activation reactions under the supervision of Dr. Chandra M. R. Volla. Upon completion of his masters, he moved to Indiana University, Bloomington for his Ph.D. research under the supervision of Prof. M. Kevin Brown. His graduate research is focusing on triplet energy transfer mediated cycloaddition reactions. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved