Org. Synth. 2025, 102, 156-184
DOI: 10.15227/orgsyn.102.0156
Synthesis of Serine Derived Oxazolines Using a Molybdenum Dioxide Catalyst
Submitted by Wyatt C. Powell, Garrett E. Evenson, and Maciej A. Walczak*
1Checked by Zengsheng Yin and Pauline Chiu
1. Procedure (Note 1)
A. Methyl ((3S,5S,7S)-adamantane-1-carbonyl)-L-serinate (1). A 1-L, two-necked, round-bottomed flask (TS 24/40, 24/40) is equipped with a 4.0 x 1.5 cm Teflon-coated, egg-shaped magnetic stir-bar, and the setup is placed inside the oven to dry (125 ℃, 4 h). The hot flask is fitted with a vacuum adapter (with stopcock and 24/40 ground glass joint) that is connected to a double manifold and secured with a Keck clip, while the other neck is stoppered with a glass stopper (TS 24/40) (Figure 1A). The flask is evacuated on high vacuum (< 1 mm Hg) and refilled with argon (three cycles) via the double manifold, then allowed to cool to rt (~25 ℃) under argon (Note 2). Under a flow of argon, the glass stopper is replaced by a powder addition funnel (TS 24/29, 75 mL) (Figure 1B), then L-serine methyl ester hydrochloride (16.0 g, 99.6 mmol) (Note 3), acetonitrile (0.50 L, 0.1 M) (Note 4), potassium carbonate (20.9 g, 149 mmol) (Note 5) are added sequentially through the funnel. After the addition is complete, the addition funnel is replaced by a glass stopper. The white suspension is stirred vigorously under magnetic stirring (750 rpm), then it is immersed in an ice/water bath (Figure 1C). Under a flow of argon, the glass stopper is replaced by a powder addition funnel (TS 24/29, 75 mL), through which 1-adamantanecarbonyl chloride (10.2 g, 49.8 mmol) (Note 6) is added in one portion. After the addition is finished, the addition funnel is replaced by a glass stopper which is secured using a Keck clip. The reaction is kept under a slightly positive pressure of argon via the double manifold. The ice bath is allowed to warm up to room temperature (~25 ℃) slowly, and the reaction mixture is stirred for 14 h (Figure 1D).

Figure 1: (A) Assembled reaction apparatus; (B) Reaction apparatus with argon flowing, in preparation for the addition of reactants and solvent; (C) Reaction is immersed in ice-water bath; (D) Reaction after stirring for 14 h. (Photos provided by checkers)
To the suspension is added deionized water (0.40 L) in one portion. The reaction mixture becomes clear and colorless (Figure 2A). The resultant mixture is transferred to a 2-L separatory funnel (Figure 2B). After separation, the aqueous phase is back-extracted with ethyl acetate (Note 7) (1 x 0.40 L, 2 x 0.20 L) and the combined organic layers are washed with 1.0 M HCl (300 mL) (Note 8), 1.0 M NaHCO3 (300 mL) (Notes 9 - 10), and saturated aqueous NaCl (300 mL) (Note 11). The organic layer is transferred into a 2-L Erlenmeyer flask and the separatory funnel is rinsed with ethyl acetate (3 x 30 mL). The combined organic layers are dried over anhydrous Na2SO4 (75 g), filtered through a 150-mL medium porosity glass fritted funnel. The Erlenmeyer flask is rinsed with ethyl acetate (3 x 30 mL) and added to the funnel. The funnel is rinsed with ethyl acetate (3 x 30 mL), and the combined filtrates are concentrated into a 1000-mL, single-necked, round-bottomed flask (TS 24/40) in three portions. The filtrate was concentrated by rotary evaporation (25 ℃, 70 torr) to yield a pale-yellow oil (Note 13) (Figure 2C).

Figure 2: (A) Reaction mixture after addition of deionized water; (B) Reaction mixture after transferring into a separatory funnel; (C) Crude product after concentration and addition of stir bar. (Photo provided by checkers)
A 4.00 x 1.50 cm Teflon-coated, egg-shaped magnetic stir-bar is added to the 1000-mL, single-necked, round-bottomed flask containing crude 1 (Figure 2C). The oil is suspended in tert-butyl methyl ether (MTBE) (100 mL) (Note 14), and the cloudy solution is magnetically stirred vigorously (1000 rpm). n-Heptane (200 mL) (Note 15) is added over 1 min to precipitate the product, and the suspension is triturated under magnetic stirring for 1 h (Figure 3A). The crystals are collected by vacuum filtration (house vacuum ~75 mm Hg) into a 150-mL medium porosity glass fritted funnel and the mother liquor is collected into a 500 mL round-bottomed flask (TS 24/40). The crystals are washed with n-heptane (50 mL), air-dried on the filter (house vacuum ~75 mm Hg, 4 h, Note 16), and further dried under high vacuum (< 1 mm Hg, 4 h) (Note 17) to afford the first crop of 1 (10.2 g) as a light, micro-crystalline powder (Figure 3B). The mother liquor was stored at -20 ℃ for 24 h, filtered; and the collected solids were washed and dried in the same manner as the first crop to afford a second crop of 1 (1.27 g) (Figure 3C). Both crops were combined to afford pure 1 (11.5 g, 82% yield, 99.5% purity) (Notes 18, 19, 20).

Figure 3: (A) Crude product 1 is treated with MTBE and n-heptane; (B) First crop of product 1; (C) Second crop of product 1. (Photos provided by the Checkers)
B. Methyl (S)-2-((3S,5S,7S)-adamantan-1-yl)-4,5-dihydrooxazole-4-carboxylate (2). A 2-L, three-necked, round-bottomed flask (TS 24/40, 24/40, 24/40) is equipped with a 5.00 x 1.75 cm, Teflon-coated, egg-shaped magnetic stir bar. The central neck is fitted with a 10-mL Dean-Stark trap (TS 24/40) topped with a water-cooled condenser (TS 24/40) that is sealed with a septum connected via a needle to an oil bubbler. One side neck is stoppered with a rubber septum that is pierced through by a temperature probe. The other side neck is fitted with a powder addition funnel (TS 24/40, Figure 4A). Through the funnel, the flask is charged with 1 (10.5 g, 99.5% purity, 37.1 mmol), MoO2(acac)2 (1.27 g, 3.71 mmol) (Note 21), 6-methyl-2-pyridinecarboxylic acid (1.04 g, 7.43 mmol) (Note 22) and toluene (1.25 L, 0.0297M) (Note 23). The funnel is replaced by a glass stopper. The flask is placed on an aluminum heating block (Note 24) and the joints are lightly greased with heavy mineral oil. The temperature probe is positioned to be immersed in the reaction mixture, and the Dean-Stark trap is wrapped with aluminum foil. (Figure 4B).

Figure 4: (A) Reaction setup before reagents and solvent are added; (B) Reaction set up before heating is started. (Photos provided by the Checkers)
The pale yellow suspension is magnetically stirred (Figure 5A) and heated to azeotropic reflux (heating probe, 112 ℃) over 30 min. The suspension turns cloudy yellow (Figure 5B). The reaction slowly turns red over the course of refluxing for 20 h (Figure 5C). Heating is stopped and the reaction is allowed to cool down to room temperature (~25 ℃) after TLC analysis showed that the reaction is complete (Figure 5D). The volume of water collected by Dean-Stark trap is about 0.8 mL (Figure 5E).
Figure 5: (A) Reaction mixture before reflux; (B) Cloudy yellow reaction mixture after refluxing for 30 min; (C) Red reaction mixture after refluxing for 20 h; (D) TLC of reaction (50% EtOAc in hexane): SM = starting material (Compound 1, Rf = 0.27), R = reaction mixture (Compound 2, Rf = 0.73); C = co-spot of R and SM, visualized by KMnO4 stain (Note 25). (E) Amount of water collected in the Dean-Stark trap after reaction is complete. (Photos provided by the checkers)
The Dean-Stark trap is removed, and the reaction mixture is transferred to a 2-L, single-necked, round-bottomed flask. The reaction mixture is concentrated by rotary evaporation (35 ℃, 50 torr to 30 torr) to ~ 0.8 L. Then the contents are transferred to a 2-L separatory funnel. The reaction mixture is washed with 1.0 M NaHCO3 (300 mL), during which a lot of flocculent precipitate developed in the biphasic mixture (Figure 6). The mixture was filtered through a pad of celite by applying house vacuum (~75 mm Hg), and the celite was washed with 100 mL EtOAc (Note 7). The filtrate collected was separated using the same 2-L separatory funnel, and the aqueous layer is back-extracted with EtOAc (2 x 200 mL) (Note 7). The combined organic layers are washed with saturated aqueous NaCl (300 mL) (Note 11), and dried over anhydrous Na2SO4 (75.0 g) (Note 12) in an Erlenmeyer flask. The mixture is filtered through a 150-mL medium porosity glass fritted funnel by applying house vacuum (~75 mm Hg), along with three EtOAc rinsings of the Erlenmeyer flask (3 x 30 mL), and concentrated into a 1-L, single-necked, round-bottomed flask (TS 24/40) in 3 portions by rotary evaporation (35 ℃, 50 torr to 30 torr), to afford a yellow oil.

Figure 6: Biphasic mixture after addition and shaking with aqueous NaHCO3. (Photo provided by the checkers)
The crude oil is quantitatively transferred to a 250-mL round-bottomed flask after diluting with EtOAc (30 mL) (Note 7), combined with EtOAc rinsings of the 1-L flask (3 x 20 mL). Then 25 g of silica gel (Note 26) is added. The contents were subjected to rotary evaporation (35 ℃, 50 mm Hg) until it became a free-flowing powder. The sample is transferred to the top of an equilibrated chromatography column (Notes 27 - 28) and eluted with 25% EtOAc in heptane (Note 29). The fractions containing product are pooled and concentrated by rotary evaporation (25 ℃, 90 torr to 30 torr), further dried on high vacuum (25 ℃, < 1 torr, 4 h) to afford 2 (8.61 g, 88% yield, 99.8% purity) as a pale yellow oil (Figure 7A), which crystallizes after storage at -20 ℃ for 24 h and then standing at room temperature (~25 ℃) overnight (Figure 7B,C) (Notes 30,31,32).
Figure 7: (A) Product 2 as an oil; (B) Product 2 after solidification; (C) Crystalline product 2. (Photos provided by the checkers)
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/about/governance/committees/chemical-safety.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
L-serine methyl ester hydrochloride,
1-adamantanecarbonyl chloride,
MoO2(acac)2,
6-methyl-2-pyridinecarboxylic acid,
potassium carbonate,
sodium bicarbonate,
sodium chloride,
anhydrous sodium sulfate,
1,3,5-trimethoxybenzene,
hydrochloric acid,
acetonitrile,
tert-butyl methyl ether,
chloroform,
toluene,
n-heptane,
ethyl acetate,
chloroform-d and
acetone-d6.
2. A Welsh vacuum pump connected to the Schlenk line was used for all high vacuum procedures, and the reaction is maintained under a positive pressure of N
2 using a Schlenk line over the course of the reaction. The checkers used argon (high purity grade, >99.995%) instead of N
2 in a double manifold, where the gas manifold is connected to an oil bubbler to prevent over-pressurization.
3.
L-Serine methyl ester hydrochloride (98%) was obtained from Combi-Blocks, Inc. which was used as received. The checkers purchased
L-serine methyl ester hydrochloride (97%) from Leyan.com Shanghai, China, which was used as received.
4.
MeCN (HPLC grade) was obtained from Fisher Chemical. The checkers obtained
acetonitrile (HPLC grade) from Fisher Scientific Korea Ltd., which was used as received.
5. The checkers purchased
potassium carbonate (99%) from Energy Chemical, which was used as received. The authors used anhydrous
K2CO3 (ACS grade) which was obtained from Fisher Chemical.
6.
1-Adamantanecarbonyl chloride (97%) was obtained from Sigma-Aldrich and was used as received. The checkers purchased
1-adamantanecarbonyl chloride (97%) from Shanghai Aladdin Bio-Chem Technology Co., Ltd., which was used as received.
7.
Ethyl acetate (ACS grade) was obtained from Fisher Chemical. The checkers purchased
ethyl acetate (GR grade) from Duksan Pure Chemicals Ltd., which was used as received.
8.
Hydrochloric acid (ACS plus grade, 12.1 M) was obtained from Fisher Chemical. The checkers purchased
hydrochloric acid (37%) from VWR Chemicals, which was diluted with water to 1.0 M for use.
9.
Sodium bicarbonate (ACS grade) was obtained from Fisher Chemical. The checkers purchased
sodium bicarbonate (AR grade), from Dieckmann (Hong Kong) Chemical Industry Co. Ltd.
10. A lot of gas is generated during the shaking and washing, so the release of pressure in the separatory funnel should be done carefully. The shaking and release of pressure should be repeated until no more gas is generated.
11.
Sodium chloride (ACS grade) was obtained from Fisher Chemical. The checkers purchased
sodium chloride (AR grade) from Dieckmann (Hong Kong) Chemical Industry Co. Ltd. to make a saturated aqueous solution.
12.
Sodium sulfate (ACS grade) was obtained from Fisher Chemical. The checkers purchased anhydrous
sodium sulfate (AR grade) from Dieckmann (Hong Kong) Chemical Industry Co. Ltd.
13. One should be careful not to concentrate for too long; otherwise, the oil would solidify and would be difficult to dissolve again.
14.
tert-Butyl methyl ether (99%) was obtained from Alfa Aesar. The checkers purchased
tert-butyl methyl ether (AR grade) from RCI Labscan Ltd., which was used as received.
15.
n-Heptane was obtained from Macron Fine Chemicals and used as received. The checkers purchased
n-heptane (GR grade) from Duksan Pure Chemicals Ltd., which was used as received.
16. The authors air-dried the compound on the filter using a water aspirator.
17. The product was placed inside a round-bottomed flask with a vacuum adapter and was dried under high vacuum until its weight became constant.
18. Analytical data for
methyl((3S,5S,7S)-adamantane-1-carbonyl)-L-serinate (
1) are as follows: mp = 100.2 - 100.6 ℃; [α]
25D = (+) 24.6 (
c 1.00, CHCl
3); IR: 3380, 2905, 2851, 1743, 1640, 1520, 1453, 1345, 1209, 1145, 1074, 977 cm
-1;
1H NMR
pdf (600 MHz, CDCl
3)
δ: 6.57 (d,
J = 6.8 Hz, 1H), 4.65 (dt,
J = 7.6, 3.8 Hz, 1H), 3.99 - 3.88 (m, 2H), 3.79 (s, 3H), 2.79 - 2.68 (m, 1H), 2.11 - 2.03 (m, 3H), 1.94 - 1.83 (m, 6H), 1.80 - 1.68 (m, 6H) ppm;
13C NMR
pdf (151 MHz, CDCl3) δ 178.9, 171.3, 64.1, 54.9, 52.9, 40.9, 39.2, 36.6, 28.2 ppm; HRMS-ESI (
m/
z) [M+Na]
+: Calcd for C
15H
23NNaO
4, 304.1519; Found 304.1519.
19. The checkers determined the purity of product
1 by quantitative
1H NMR
pdf analysis using
1 (23.0 mg, 0.0817 mmol) and
1,3,5-trimethoxybenzene (Sigma-Aldrich
TraceCERT
® Standard for quantitative NMR, 13.5 mg, 0.0803 mmol) in CDCl
3.
20. A second reaction performed by the checkers on the same scale provided 11.4 g (98.2% purity, 80% yield) of
1.
21.
MoO2(acac)2 (99%) was obtained from Alfa Aesar and was used as received. The checkers purchased
MoO2(acac)2 (>95%) from TCI, which was used as received.
22.
6-Methyl-2-pyridinecarboxylic acid (>95%) was obtained from Matrix Scientific and was used as received. The checkers purchased
6-methyl-2-pyridinecarboxylic acid (>98%) from TCI, which was used as received.
23.
Toluene (ACS grade) was obtained from Sigma-Aldrich and was used as received. The checkers purchased
toluene (ACS grade) from Duksan Pure Chemicals Ltd., which was used as received.
24. The checkers used an aluminum heating block (designed for 2000 mL round-bottomed flask), which was purchased from Beijing Synthware Glass Inc. The authors used a heating mantle from Glas-Col (catalogue No. 0410), Watts = 500, Volts = 115, 18 cm bottom diameter, 22 cm top diameter, and 10 cm height. The transformer used was a Staco Energy Products Co. variable auto transformer (model No. 3PN101OB) set to 70% maximum output voltage, Input 120 V 50/60 Hz, Output 0-140 V, 10 AMP, 1.4 KVA, 12 AMP fuse.
25. The checkers performed TLC analysis on TLC Plates (Silica gel 60 F254, Aluminum Sheets, from Supelco) in 50%
EtOAc in hexane, by visualization with
KMnO4 stain. The authors performed TLC analysis on SiliCycle TLC plates in 50%
EtOAc in heptane.
26. Silica gel was purchased from SiliCycle (SiliaFlash P60, particle size: 40-63 μm, pore size 60 Å) and was used as received. The checkers purchased Silica Gel 60 (particle size 0.040-0.063 nm) from Millipore, Sigma-Aldrich Canada, which was used as received.
27. A 2-L column (8 cm diameter) is charged with 200 g of silica gel (10.5 cm high). It is equilibrated by flushing 1 L of
n-heptane through the silica gel repeatedly (5 flushes) until no air remains. The checkers used a column with a size of 80 x 305 mm (Figure 8).
Figure 8: Column after sample is loaded and sand is added (Photo provided by the checkers)
28. The loaded silica gel is transferred to the column, and the flask is washed with heptane (3 x 20 mL) and
EtOAc (3 x 20 mL) into the column. The column is equilibrated again with heptane (1 flush), and the column is charged with sand (100 g). The checkers, after transferring the dry loaded silica gel to the column, washed the flask with
dichloromethane (3 x 20 mL) and transferred this to the column. Subsequently the column is equilibrated with heptane, and the top of the column is covered with a layer of sand (100 g).
29. The authors eluted the column with 25%
EtOAc/heptane (3.0 L) and fractions are collected into 140 18 x 150 mm test tubes. TLC analysis of the fractions in 50%
EtOAc/heptane visualized with
KMnO4 stain indicated the fractions containing product. The checkers eluted the column with 25%
EtOAc/heptane (3.0 L) into 18 x 150 mm test tubes.
KMnO4 staining showed that fractions 37 - 124 contained product
2 (Figure 9).
Figure 9: TLC analysis of fractions. stained with KMnO4 stain (Photo provided by the checkers)
30. Analytical data for
methyl (S)-2-((3S,5S,7S)-adamantan-1-yl)-4,5-dihydrooxazole-4-carboxylate (
2). mp = 44.7 - 45.7 ℃; [α]
25D = (+) 114.2 (
c 1.16, CHCl
3); IR: 2905, 2851, 1742, 1646, 1453, 1351, 1322, 1275, 1206, 1180, 1056, 970 cm
-1;
1H NMR
pdf (600 MHz, CDCl
3)
δ: 4.69 (dd,
J = 10.5, 7.8 Hz, 1H), 4.42 (dd,
J = 8.7, 7.8 Hz, 1H), 4.34 (dd,
J = 10.5, 8.7 Hz, 1H), 3.77 (s, 3H), 2.07 - 1.97 (m, 3H), 1.95 - 1.85 (m, 6H), 1.76 - 1.68 (m, 6H) ppm;
13C NMR
pdf (151 MHz, CDCl
3) δ 176.4, 172.2, 69.2, 68.1, 52.7, 39.6, 36.6, 35.5, 28.0 ppm; HRMS-ESI (
m/
z) [M+H]
+: Calc'd for C
15H
22NO
3, 264.1594; Found 264.1594.
31. The checkers determined the purity of product
2 by quantitative
1H NMR
pdf analysis using
2 (24.2 mg, 0.0919 mmol) and
1,3,5-trimethoxybenzene (Sigma-Aldrich
TraceCERT
® Standard for quantitative NMR, 13.8 mg, 0.0821 mmol) in CDCl
3.
32. A second reaction performed by the checkers on the same scale provided 8.73 g (98.5% purity, 89% yield) of
2. A second reaction of the same scale performed by the authors provided 8.77 g (89% yield) of
2.
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Oxazolines and thiazolines are common motifs in many peptide derived natural products.
2, 3, 4 These products include thiopeptide antibiotics
5 and metal chelating siderophores.
6, 7 Biosynthetically, azolines are installed from ribosomally synthesized peptides containing serine, threonine, and cysteine by cyclodehydratase enzymes.
8 The post-translational modification allows organisms to diversify peptides' structure and function. In organic synthesis, this idea can be expanded to include further functionalizations of azolines. They can undergo oxidation, producing oxazoles and thiazoles,
9, 10 altering their structures and making the products more resistant to hydrolysis. Oxazolines can also serve as activated amides, which can form thioamides or amines under mild conditions through reactions of nucleophiles at the C2 position.
11, 12 Oxazolines form carbon heteroatom bonds at C5 with chalcogens, halogens, azides, and other nucleophilic reagents under acidic or basic conditions,
13, 14 such as oxazoline
3 that opens with PhSH to form Nelfinavir.
15, 16, 17 The azoline can be used as an intermediate in the enolization of the C2 exomethine position. Installation of an azoline also provides a way to differentiate the nitrogen atom of multiple amides, providing access to N-functionalized peptides. This is demonstrated in the synthesis of α-cytidine from ribose-aminooxazoline
4.
18 Therefore, methods to produce azolines can enable the editing of peptides and the differentiation of natural products and biologically relevant molecules.
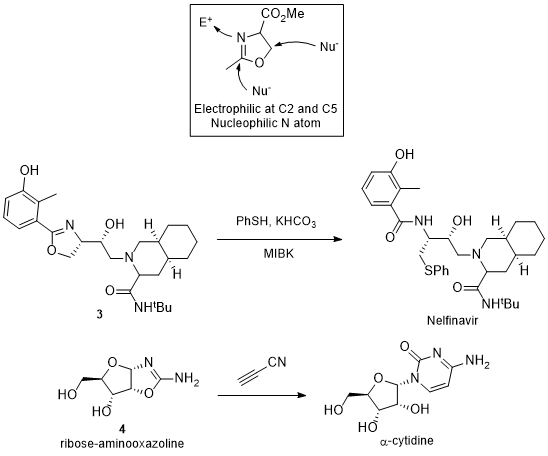
Scheme 1: Azolines as Reactive Intermediates
Various methods to synthesize azolines exist
19 but those that use β-hydroxyamides or β-mercaptoamides as the substrate are the most practical because these cyclodehydration reactions can be applied on accessible serine, threonine, and cysteine derivatives. Additionally, if an azoline is needed as a product, rather than as an intermediate, it is beneficial to install the azoline late in synthesis due to potential issues that can arise due to the relative reactivity of an azoline such as hydrolysis or epimerization of the C2-exocyclic position of the heterocycle.
Two strategies can be employed to achieve dehydrative cyclizations of cysteine, serine and threonine derivatives (Scheme 2A). The first involves the activation of the alcohol, and the second involves the activation of the amide. Alcohol activation requires the amide oxygen (or thioamide sulfur) atom to displace the alcohol. This is typically achieved with a dehydrating agent. These dehydrating agents include DAST,
20, 21 Deoxo-Fluor,
20 XtalFluor-E,
21, 22 the Burgess reagent,
23, 24 DIC/CuOTf
25 and DEAD/Ph
3P
26, 27, 28 in the Mitsunobu reaction. These reactions can typically achieve high yields; however, they are plagued with several issues including functional group tolerance, the unwanted formation of aziridines (most often observed in the functionalization of threonine),
28 and the production of potentially toxic byproducts that require extensive purification and waste disposal. Another drawback of this approach is the inversion of stereochemistry that occurs at C5 of the oxazoline, and the need for thioamide installation before thiazoline synthesis.
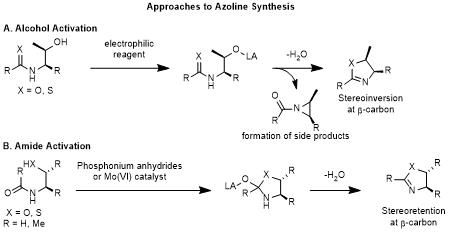
Scheme 2: Chemical Approaches to Azoline Synthesis
Amide activation is an alternative approach, where the amide is activated as the electrophile and the alcohol or thiol displaces the amide oxygen atom (Scheme 2B). This approach does not require thioamides for thiazoline synthesis and it preserves stereochemistry of threonine derived 5-methyloxazolines. Amide activations are of particular interest because this transformation can be applied to simple derivatives of natural serine, threonine, and cysteine derivatives to obtain both oxazoline and thiazoline heterocycles. One method that uses this mechanistic strategy is with phosphonium anhydrides, such as Hendrickson's POP reagent.
29 Vorbrüggen observed that (Ph
3P)
2O·2TfO
- activates the amide of
5 to form oxazoline
6a or thiazoline
6b in the presence of a tertiary amine base.
30 Loughlin and coworkers developed a cyclic phosphonium anhydride
7 and used trityl protected substrates
5c and
5d to inhibit unproductive alcohol/thiol activation, while the trityl protective group is cleaved in situ by liberated triflic acid.
31 Then, cyclization likely proceeds through a 5-endo-dig mechanism (Scheme 3).
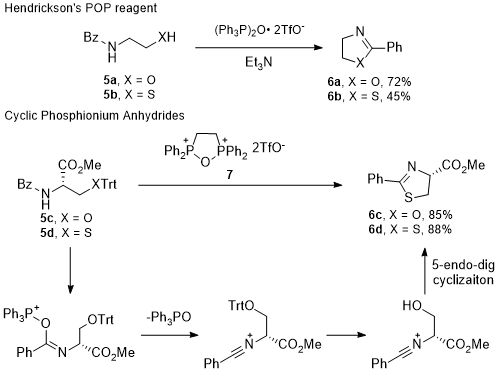
Scheme 3. Phosphonium Anhydrides in the Synthesis of Azolines
In addition to electrophilic phosphonium dehydrating agents, Ishihara and coworkers discovered that molybdenum oxides display good catalytic activities in cyclodehydration of serine
8a, threonine
8b, and cysteine
8c derivatives (Scheme 4).
32 The reaction conditions require a catalytic quantity of the metal and they produce water as the only waste product that is removed through azeotropic reflux. The reaction of dipeptides
8 with either catalysts
C1 or
C2 in refluxing toluene produced
9 in 87% and 68% yield, respectively. Molybdenum (VI) dioxides promoted cyclization with stereoretention at the side chain of threonine and producing thiazolines with cysteine derivatives, indicating these reactions proceeded under the amide activation mechanism. Serine dipeptide
9a was cyclized without epimerization at the C2-exomethine position, but the synthesis of 5-methyloxazoline
9b and thiazoline
9c displayed some level of epimerization (5% and 15% respectively). Ishihara and coworkers later demonstrated additional control of the cyclization by modulating the Lewis and Brønsted basicity of the catalysts with N,O-chelating bidentate ligands (catalysts
C3 and
C4), which significantly reduced the epimerization of thiazoline (1:99) and 5-methyloxazoline (94:6).
33
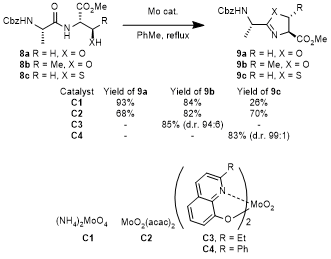
Scheme 4. Molybdenum Oxides in the Synthesis of Azolines
The Mo (VI) method was utilized in the synthesis of a 2-hydroxyphenyl oxazoline moiety of antitumor agent BE-70016 (Scheme 5).
34 Exposure of
10 to the previously optimized conditions using MoO
2(acac)
2 (
C1) or (NH
4)
2MoO
4 (
C2) afforded
11 in 5% and 17% yield, respectively. The authors rationalized the product was likely forming a stable chelate
12, which was catalytically inactive. Addition of pentafluorobenzoic acid improved the yield of
11 to 76%. The carboxylic acid additive likely facilitates the conversion of
12 to reactivate the catalyst.
Scheme 5. Synthesis of Antitumor Substance BE-70016
Walczak and coworkers utilized the Mo(VI) system for the total synthesis of micrococcin P1 and thiocillin I.
35 However, cyclization of
13 with MoO
2(acac)
2 (
C1) did not proceed to completion, likely due to the low solubility and deactivating aggregation of MoO
2(acac)
2 (Scheme 6). Picolinic acids were predicted to improve solubility and stability of the catalyst, in addition to modulating its electrophilic character and sterics. The addition of
15 as an additive, predicted to from complex
16 in situ, greatly improved the yield of
14. 6-Methylpicolinic acid was determined to be a highly effective additive in the synthesis of thiazoline
14.
Scheme 6. Picolinate Ligands in Molybdenum Dioxide Catalysis - Cyclization of Cysteine Derivatives
The utility of catalyst
16 was also explored in the synthesis of oxazolines (Scheme 7).
36 It was determined that precomplexation of the catalyst was beneficial to achieve high yields of oxazoline
18, as well as to suppress the formation of the dehydroalanine elimination product. Additional ligands were successful in eliminating the unproductive elimination but produced oxazolines in lower yields. Of particular interest are the less basic thiazole and oxazole containing ligands, which produced
18 in acceptable yield, demonstrating that the basicity of the neutral ligand, which is thought to assist in proton transfer, does not play a significant factor in the success of the reaction. The catalyst was able to produce azolines in acceptable to high yield that contained sensitive functional groups, such as Fmoc-protected amines (
21-
22,
27-
31). In cyclizations with substrates containing highly coordinating primary amide and hydroxamate functional groups (
24,
32), the catalyst remained active, despite the possibility of catalyst deactivation through ligand exchange with the substrate or product.
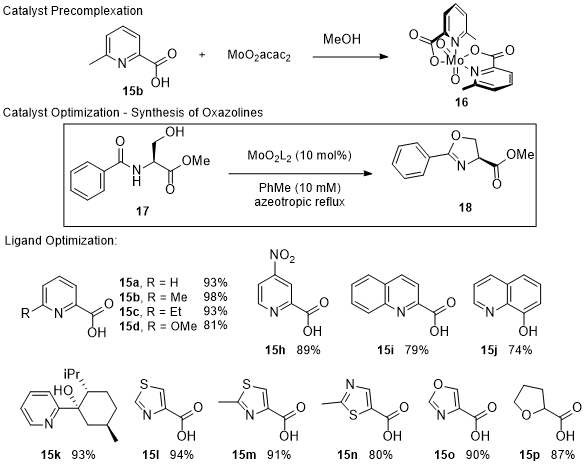
Scheme 7. Picolinate Ligands in Molybdenum Dioxide Catalysis - Cyclization of Serine Derivatives
Scheme 8: Picolinate Ligands in the Molybdenum-catalyzed Synthesis of Oxazolines and Thiazolines
While the MoO
2(6-MePic)
2 catalyst is more resistant to deactivation than MoO
2(acac)
2, it is not indefinitely stable under the reaction conditions likely due to aggregation and polymerization. Therefore, a tetradentate ligand
35 was invented that extended the lifetime of the catalyst, remaining active over reaction times of several days (Scheme 9).
36, 37 The catalyst was isolated by chromatography and could be recovered from the reaction mixtures after the cyclization is complete. This improved stability enabled cyclizations of natural products and pharmaceuticals, including functionalizations of strained cyclic peptides. This includes the in-situ reaction of the oxazoline intermediate to introduce side-chain and backbone modifications to cyclic peptides.
Scheme 9. Stabilizing Tetradentate Ligands in Molybdenum Dioxide Catalysis - Synthesis of Azolines
Taken together, 6-MePic additive offers the most optimal catalytic system for the installation of azolines in peptides and complex substrates taking into consideration practical aspects and the cost. Our protocol utilizes the 6-MePic catalyst which is generated in situ for improved performance and stability. Cyclization of 1 with MoO2(acac)2 afforded 2 in 60% isolate yield, and the addition of 6-MePic increased the yield of 2 to 89%. Cyclodehydration of 1 with 6-MePic as an additive after 3h afforded 2 in 74% yield, and the yield increased to 89% after 18 h, which demonstrates that the ligand helps keep the catalyst active over longer reaction times. A limitation of the method is that it must be conducted under high dilution conditions. We found the concentration could be increased to 30 mM with no deterioration of the yield. The procedure above offers a mild synthesis of oxazolines and thiazolines, while avoiding the use of harsh electrophilic reagents and that produces water as the only waste product.
Appendix
Chemical Abstracts Nomenclature (Registry Number)
6-Methyl-2-pyridinecarboxylic acid; (934-60-1);
Acetonitrile; (75-05-8);
Adamantanecarbonyl chloride; (2094-72-6);
Bis(acetylacetonato)dioxomolybdenum; (17524-05-9);
Ethyl acetate; (141-78-6);
Heptane; (142-82-5);
Hydrochloric acid; (7647-01-0);
L-Serine, methyl ester, hydrochloride; (5680-80-8);
Methyl ((3S,5S,7S)-adamantane-1-carbonyl)-L-serinate: N-(tricyclo[3.3.1.13,7]dec-1-ylcarbonyl)-L-serine methyl ester; (1)(1369391-08-2);
Methyl (S)-2-((3S,5S,7S)-adamantan-1-yl)-4,5-dihydrooxazole-4-carboxylate: methyl (4S)-4,5-dihydro-2-tricyclo[3.3.1.13,7]dec-1-yl-4-oxazolecarboxylate; (2) (369391-11-7);
Potassium carbonate; (584-08-7);
Sodium bicarbonate; (144-55-8);
Sodium chloride; (7647-14-5);
Sodium sulfate; (7757-82-6);
tert-Butyl methyl ether (1634-04-4);
Toluene; (108-88-3);
Water; (7732-18-5);

|
Maciej Walczak received his undergraduate degree (maxima cum laude) from Adam Mickiewicz University in Poznan, Poland. In 2003 he started his graduate studies at the University of Pittsburgh, where he worked on the synthesis and reactions of strained carbocycles. After graduating in 2009, he moved to New York as a Brodeur Foundation postdoctoral fellow at Memorial Sloan-Kettering Cancer Center, where he was involved in the synthesis of glycoproteins, anticancer vaccines, and antimetastatic agents. In August 2013 he started his independent career at the University of Colorado Boulder as an Assistant Professor of Chemistry and was promoted to Associate Professor in 2020. |

|
Wyatt Powell studied chemistry and biochemistry at the University of Colorado Boulder as an undergraduate student. In 2018, Wyatt began a PhD at the University of Colorado Boulder. His research focuses on a molybdenum catalyzed cyclodehydration reaction, and chemical synthesis of the microtubule associated protein tau. |

|
Garrett Evenson attended the University of Montana where he worked as an undergraduate researcher in the synthesis of high oxidation state transition metal complexes for CH bond abstraction. Then, in 2019, he began his graduate studies in organic chemistry at the University of Colorado Boulder under the advisement of Dr. Walczak. |

|
Zengsheng Yin obtained his Ph.D. from Donghua University in Shanghai in 2010, under the supervision of Prof. Feng-Ling Qing. He joined Prof. Pauline Chiu's group as a postdoctoral researcher in 2013 and a senior research fellow in 2020, working on the total synthesis of natural products. |
Copyright © 1921-2025, Organic Syntheses, Inc. All Rights Reserved