Org. Synth. 2025, 102, 185-202
DOI: 10.15227/orgsyn.102.0185
Synthesis of 1-(phenylsulfonyl)bicyclo[1.1.0]butane from Methyl Phenyl Sulfone and Epichlorohydrin
Submitted by Joanna E. Muir, Myunggi Jung, and Vincent N. G. Lindsay*
1Checked by Yu-Che Chang and M. Kevin Brown
1. Procedure (Note 1)
1-(Phenylsulfonyl)bicyclo[1.1.0]butane (1). A 500-mL, oven-dried, single-necked (24/40), round-bottom flask is charged with methyl phenyl sulfone (6.00 g, 38.4 mmol, 1.00 equiv) (Note 2) and equipped with a 16 x 38 mm, egg-shaped, Teflon-coated magnetic stir bar, a rubber septum (24/40), and a nitrogen inlet connected to a Schlenk line (Figure 1A). The flask is held under vacuum for 10 min, then slowly backfilled with nitrogen gas (Note 3), and the operation was performed in triplicate. The setup is maintained under a slight positive pressure of nitrogen gas. Anhydrous tetrahydrofuran (154 mL, 0.25 M) (Note 4) is added to the flask using a 60-mL syringe, and vigorous stirring (750 rpm) is initiated. Once the methyl phenyl sulfone is fully dissolved (ca. 5 min) resulting in a colorless solution (Figure 1B), the reaction is submerged in a -78 ℃ cooling bath (acetone/dry ice) (Note 5) and allowed to cool for 10 min. Di-n-butyl magnesium (n-Bu2Mg) (42.7 mL, 38.4 mmol, 1.00 equiv) (Notes 6 & 7) is slowly added in two successive portions to the flask over 5 min using a 24-mL syringe, and the reaction is stirred at -78 ℃ for 30 min (Figure 1C). At this point, formation of a white crystalline precipitate is observed. Epichlorohydrin (3.01 mL, 38.4 mmol, 1.00 equiv) (Note 8) is added to the reaction vessel in one portion using a 6-mL syringe. The cooling bath is removed, the reaction is allowed to warm to room temperature (~25 ℃) and stirred for 3 h. Completion of the first stage is confirmed by TLC analysis using hexanes : EtOAc (2 : 1) as eluent (Note 9), at which point the reaction is a translucent pale-green solution containing small amounts of solid sediments (Note 10) (Figure 1D).

Figure 1. A. Setup for procedure; B. Appearance of the reaction after dissolution of methyl phenyl sulfone; C. Reaction submerged in cooling bath 30 min after the addition of n-Bu2Mg; D. Appearance of the reaction 3 h after the addition of epichlorohydrin
The reaction is cooled to 0 ℃ (ice/water bath) (Figure 2A), and benzenesulfonyl chloride (6.40 mL, 49.9 mmol, 1.30 equiv) (Note 11) is slowly added using a 6-mL syringe over 2 min, affording a pale-yellow homogenous solution (Figure 2B). The reaction is stirred for an additional 10 min at 0 ℃ before the cooling bath (ice/water) is removed. The mixture is allowed to stir at room temperature (~25 ℃) for 18 h, resulting in a homogenous yellow solution (Figure 2C). Completion of the second step is monitored by TLC analysis using hexanes : EtOAc (2 : 1) as eluent (Note 12).
Figure 2. A. Appearance of the reaction before addition of benzenesulfonyl chloride; B. Appearance of the reaction immediately after the addition of benzenesulfonyl chloride; C. Appearance of the reaction 18 h after addition benzenesulfonyl chloride
The reaction is cooled to -78 ℃ for 10 min using a dry ice/acetone cooling bath (Note 5), before n-butyllithium (n-BuLi) (19.2 mL, 46.3 mmol, 1.20 equiv) (Note 13) is added (at -78 ℃) dropwise in two successive portions (over 20 min overall) using a 24-mL syringe (Figure 3A). The reaction is stirred for an additional 30 min at -78 ℃. The cooling bath is removed, and the reaction is stirred at room temperature (~25 ℃) for 3 h (Note 14), resulting in a homogenous orange solution (Figure 3B). Completion is monitored by TLC analysis using hexanes : EtOAc (2 : 1) (Note 15).
Figure 3. A. Appearance of the reaction immediately after addition of n-BuLi; B. Appearance of reaction 3 h after the cooling bath is removed.
The reaction is quenched while stirring with a "half saturated" aqueous NH4Cl solution (150 mL) (Figure 4A) (Note 16). The resulting biphasic mixture is transferred to a 500-mL separatory funnel, rinsing the reaction flask twice with 30 mL of EtOAc. The organic layer is collected, and the aqueous layer is further extracted twice with EtOAc (100 mL). The organic layers are combined into a 1-L Erlenmeyer flask, then dried by addition of one portion of MgSO4 (10 g) followed by manual stirring for 10 seconds. The mixture is filtered using a sintered glass funnel (350-mL, medium grit) fitted to a 1-L round-bottomed flask, while rinsing with additional EtOAc (50 mL). The organic solvent is evaporated under gradually reduced pressure (300 to 25 mbar at 25 ℃) until approximately 50 mL of solution remained. Silica (8 g) is added to the resulting solution (Note 17). The mixture is returned to the rotatory evaporator and concentrated under reduced pressure (25 ℃, 300 to 25 mbar) to a powdery solid (Figure 4B). The powdery solid is further dried under high vacuum for 30 min. The crude solid is then subjected to flash chromatography on silica gel (Notes 17 & 18) using EtOAc (Note 19) and hexanes (Note 20), to furnish 1-(phenylsulfonyl)bicyclo[1.1.0]butane (1) (4.2 g, 56% yield, 99.7% purity) as a white crystalline solid (Notes 21 & 22) (Figure 4C).

Figure 4. A. Appearance of the reaction after quenching with H2O and NH4Cl; B. Appearance of powdery residue after concentrating with silica; C. Appearance of final product (1) after purification
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/about/governance/committees/chemical-safety.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
methyl phenyl sulfone,
di-n-butylmagnesium (1.0 M),
benzenesulfonyl chloride,
n-butyllithium (2.5 M),
tetrahydrofuran,
epichlorohydrin,
EtOAc,
magnesium sulfate, deionized water, hexanes,
ammonium chloride (aqueous), silica gel,
deuterated chloroform, as well as the proper procedures for working under inert atmosphere, solvent evaporation, working with dry ice baths, the application of high vacuum.
2.
Methyl phenyl sulfone (>98%) was purchased from AmBeed Inc. and used as received.
3.
Nitrogen gas was purchased from ARC3 Gases (NI300, High Pressure Industrial Grade) and used as purchased.
4. Anhydrous
tetrahydrofuran (inhibitor-free, ≥99.9%) was purchased from Sigma Aldrich and used as received.
5. For dry ice/acetone cooling bath, slowly add acetone to dry ice. During the cooling cycles, dry ice is added as needed to keep the bath temperature at -78 ℃.
6.
Di-n-butyl magnesium (1M in
heptane, Sure/Seal
TM packaging system) was purchased from Sigma Aldrich and titrated prior to use in triplicate, and the concentration of the bottle used was found to be 0.90 M. The age and concentration of this reagent was found to be crucial to get reproducible results, as older batches of reagents with a concentration of 0.50 M or less led to a significant decrease in yield. Titrations were conducted according to the method of Knochel.
2 Lithium chloride (99%, anhydrous) used for the titration was purchased from Acros Organics and used as received; iodine (resublimed crystals, 99.8%, certified ACS) was purchased from Alfa Aesar and used as received.
7. When available for purchase,
di-n-butyl magnesium (1M in
Et2O/hexanes, Sure/Seal
TM packaging system) can also be employed for the same procedure and generally affords higher yields (roughly a 15% increase).
8.
Epichlorohydrin (>99.0%) was purchased from TCI Chemicals and used as received.
9. The reaction progress is evaluated using TLC analysis using glass-backed silica plates, developed with 2:1 hexanes/
EtOAc and visualized under UV light (254 nm). The R
f of the
methyl phenyl sulfone is 0.50, and the R
f of intermediate
A (protonated form, 3-(phenylsulfonyl)cyclobutanol) is 0.10.
Figure 5. Thin layer chromatography (TLC) analysis of Step 1 (3 h after epichlorohydrin addition) to intermediate A (Note 9)
10. At this point of the reaction, the color may appear pale green with white solids, other times the solid may have more of a green hue. Either appearance is acceptable. Check TLC for confirmation of reaction completion.
11.
Benzenesulfonyl chloride (>99.0%) was purchased from TCI Chemicals and used as received.
12. TLC analysis is performed according to
Note 9; intermediate
A (protonated form, 3-(phenylsulfonyl)cyclobutanol) has R
f = 0.10 and intermediate
B (3-(phenylsulfonyl)cyclobutylbenzenesulfonate) has R
f = 0.30.
Figure 6. Thin layer chromatography (TLC) analysis of Step 2 (18 h after PhSO2Cl) to intermediate B (Note 12)
13.
n-Butyllithium (2.5 M in hexanes, Sure/Seal
TM packaging system) was purchased from Sigma-Aldrich and titrated prior to use, and the concentration of the bottle used was found to be 2.41 M. Titrations were conducted according to the method of Kofron.
3 Diphenylacetic acid (99%) used for the titration was purchased from Thermo Scientific and recrystallized from anhydrous
toluene, vacuumed and dried prior to use.
14. On smaller scale (1.0 g of
methyl phenyl sulfone), the last step is often completed in 2 h when monitored by TLC analysis.
15. TLC analysis is performed according to
Note 9, intermediate
B has R
f = 0.30, and
1-(phenylsulfonyl)bicyclo[1.1.0]butane (
1) has R
f = 0.76. If
benzenesulfonyl chloride is prominent to any degree on developed plate, the TLC can be developed in 9:1 hexanes/
EtOAc.
Figure 7. Thin layer chromatography (TLC) analysis of Step 3 (3 h after addition of n-BuLi) to Product 1 (Note 15)
16. A 'half saturated' aqueous solution of
NH4Cl is prepared by diluting 75 mL aq. sat.
NH4Cl with 75 mL
H2O.
17. Silica gel (SiliCycle
TM SiliaFlash
TM F60, 40-63 μM, 60 Å) was purchased from SiliCycle
TM and used as received.
18. Purification by column chromatography is performed using 140 g of dry silica gel (
Note 17) in a 500 mL, 5 cm diameter, 24/40 glass column. The column is plugged with cotton and topped with 2 cm (height) of sand. The silica is pre-wet using 5%
EtOAc (
Note 19) in hexanes (
Note 20), and the crude product is dry loaded onto the silica. Sand (2 cm height) (
Note 23) is added to ensure a level silica gel line during loading. A gradient solvent system is employed, eluting first with 1000 mL of 5%
EtOAc in hexanes, followed by 2000 mL of 8%
EtOAc in hexanes gradually added during elution. Fractions of 100 mL are collected, totaling 35 fractions. The desired product is obtained in fractions 21-31, and are combined into a 2000 mL, single-necked (24/40) round bottom flask. The bulk of the solvent is removed by rotatory evaporation (300 to 25 mbar at 25 ℃). The remaining solution (
ca. 50 mL) is transferred to a 250 mL, single necked (24/40), round-bottom flask, rinsing with
EtOAc (20 mL). The residual solvent is removed by rotatory evaporation (300 to 25 mbar at 25 ℃) to furnish the product. The product is further dried under high vacuum (
ca. 0.1 torr) at ambient temperature for 4 h.
19.
Ethyl acetate (
EtOAc, ≥99.5%, Certified ACS) was purchased from Fischer Scientific and used as received.
20. Hexanes (≥98.5%, Certified ACS) was purchased from Fischer Scientific and used as received.
21. The product (
1) possesses the following properties: R
f = 0.76 (2 : 1 hexanes :
EtOAc); mp 75-77 ℃;
1H NMR
pdf (500 MHz, CDCl
3) δ 7.99 - 7.87 (m, 2H), 7.64 - 7.59 (m, 1H), 7.56 (t,
J = 7.5 Hz, 2H), 2.57 (p,
J = 2.8 Hz, 1H), 2.52 (d,
J = 3.8 Hz, 2H), 1.39 (d,
J = 2.2 Hz, 2H);
13C NMR
pdf (126 MHz, CDCl
3) δ 142.0, 133.2, 129.3, 127.2, 38.4, 23.2, 12.7; IR (neat) 3092, 2979, 1445, 1303, 1287, 1156, 1146, 1112, 1082, 1073, 818, 754. HRMS, [M] Calc'd for C
10H
10O
2S: 194.0402. Found: 194.0399. Purity was determined to be 99.7 wt% by quantitative
1H NMR
pdf using 18.3 mg of (
1) and 14.0 mg of
1,3,5-trimethoxybenzene (99%) (purchased from Combi-Blocks Chemicals and used as received).
22. A second run on identical scale yielded product
1 (4.4 g, 59% yield) in >99.9 wt% purity determined by quantitative
1H NMR (
Note 21). In that case, the
benzenesulfonyl chloride reagent was purified via filtration through a basic alumina (
Note 24) plug (6 cm long) as a solution in anhydrous
Et2O, followed by concentration, prior to use in the reaction.
23. Sand (sea-washed) was purchased from Fischer Scientific and used as received.
24.
Aluminum oxide, for chromatography, basic, Brockmann I, 40-300 μm, 60 Å was purchased from Thermo Fischer Scientific and used as received.
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Functionalized cyclobutane derivatives constitute ubiquitous structural motifs commonly found in bioactive compounds, natural products and specialized materials.
4 In addition to classical [2+2] cycloaddition and ring-closing strategies, an increasingly popular approach to their formation involves the functionalization of activated bicyclo[1.1.0]butane (BCB) derivatives via cleavage of their bridging bond.
5 Such a process generally results in a significant release of strain, hence these substrates are often referred to as 'strain-release' reagents, where the presence of an electron-withdrawing group at the bridgehead position considerably accelerates the reaction. 1-Sulfonylbicyclo[1.1.0]butanes (
e.g.,
1) constitute a powerful subset of such reagents and have found widespread utility in this regard, where the resulting sulfonyl group serves as a handle for further functionalization (Figure 8).
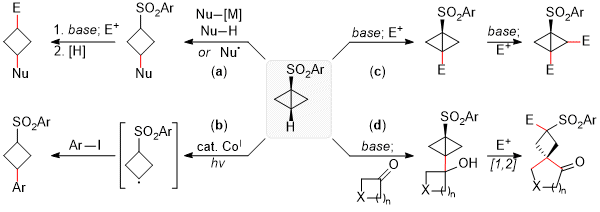
Figure 8. Utility of 1-sulfonylbicyclo[1.1.0]butanes as spring-loaded intermediates in organic synthesis
For example, the addition of nucleophiles (Nu = H, alkyl, alkenyl, NR
2, SR, SeR, N
3) readily proceeds in mild conditions, affording 3-sulfonylcyclobutyl substituents that can be further elaborated via alkylation and reduction events (Figure 8a).
6 Treatment with a cobalt(I) catalyst instead under light irradiation leads to the initial formation of a Co(III)-cyclobutyl intermediate susceptible to homolysis, affording a cyclobutyl radical which can be functionalized in the presence of aryl iodides, Michael acceptors or acylating agents (Figure 8b).
7 The distinct acidity of C-H bonds present in BCBs and the directing effect of the sulfonyl group allows for the regioselective introduction of various substituents via sequential deprotonation and treatment with electrophiles or coupling partners, leading to highly substituted 1-sulfonylbicyclo[1.1.0]butanes (Figure 8c).
8 When the electrophile is a cyclic ketone, various spirocyclic cyclobutanes can be readily generated through a subsequent semipinacol rearrangement in the presence of acids or electrophiles (Figure 8d).
9The first general approach to 1-sulfonylbicyclo[1.1.0]butanes was reported in a seminal report from Gaoni (Figure 9a).
10 Deprotonation of an aryl methyl sulfone using ethylmagnesium bromide and electrophilic quench with allyl bromide provides a homoallyl sulfone, which is then oxidized to the γ,δ-epoxy sulfone using
m-CPBA. Deprotonation of this sulfone with
n-BuLi leads to a cyclopropylcarbinol via intramolecular nucleophilic substitution. Activation of the resulting alkoxide using MsCl followed by subsequent deprotonation/cyclization with
n-BuLi leads to the corresponding 1-sulfonylbicyclo[1.1.0]butane. The procedure was later modified by Baran and co-workers, where the key homoallyl sulfone intermediate was accessed via reduction of a sulfonyl chloride followed by treatment with 4-bromo-1-butene (Figure 9b).
6d,eFigure 9. Previous synthetic routes to 1-sulfonylbicyclo[1.1.0]butanes
In 2022, our research group reported a streamlined protocol where the desired 1-sulfonylbicyclo[1.1.0]butanes can be accessed in a single pot from methyl sulfones and inexpensive epichlorohydrin via the formation of a 3-sulfonylcyclobutanoxide intermediate (present report and Figure 10).
11,12Figure 10. Selected substrate scope of 1-sulfonylbicyclo[1.1.0]butanes (performed on a 0.5 mmol scale of the methyl sulfone)
The main advantage of this approach comes from its succinct nature and the fact that all reagents needed are commercially available for most scope entries. In cases where the corresponding methyl sulfone is not commercially available, it can readily be prepared in one step by thioanisole oxidation
13 or Cu-catalyzed cross-coupling of aryl iodides with NaSO
2Me.
14 In addition to the title compound
1, a wide variety of sulfonyl-substituted BCBs were readily accessed using a similar protocol as the one described herein, including alkyl (
12-
14) and heteroaryl sulfones (
15-
17) as well as a sulfonamide (
18). Moreover, substituted
epichlorohydrin derivatives were reported as suitable reagents, leading to the direct production of bridgehead- (
19) and bridge-substituted (
20) BCBs.
Appendix
Chemical Abstracts Nomenclature (Registry Number)
Methyl Phenyl Sulfone; (3112-85-4)
Epichlorohydrin; (106-89-8)
Di-n-butylmagnesium; (1191-47-5)
Benzenesulfonyl Chloride; (98-09-9)
n-Butyllithium; (109-72-8)

|
Joanna E. Muir received her B.S. in Biochemistry in 2020 from Florida Gulf Coast University, where she worked under the mentorship of Prof. Gregory Boyce on efforts towards the synthesis of cyclopropenone-containing natural products. She started her graduate studies in the Lindsay Lab at North Carolina State University in 2021, where she is currently working on the development of new synthetic methods involving strained organic compounds. |

|
Myunggi Jung received his B.S. in Pharmaceutical Sciences in 2012 from Yeungnam University and his M.S. in Medicinal and Pharmaceutical Chemistry in 2014 from Seoul National University. From 2014 to 2018, he worked in medicinal chemistry at Daewoong Pharmaceutical. He obtained his Ph.D. in Chemistry from North Carolina State University in 2023, working with Prof. Vincent Lindsay on the chemistry of strained rings. After a postdoctoral training at The University of North Carolina, Chapel Hill (Alexanian lab, Department of Chemistry), he began his independent career at Yeungnam University (College of Pharmacy) in 2024, where he is currently an Assistant Professor. |

|
Vincent N. G. Lindsay received his B.Sc. in Chemistry in 2007 from Université de Montréal. He pursued his doctoral studies with Prof. André B. Charette (Université de Montréal) and received his Ph.D. in Chemistry in 2013, working on the development of asymmetric Rh-catalyzed cyclopropanation reactions. He then worked as a FRQNT postdoctoral fellow at the University of California, Berkeley under the supervision of Prof. Richmond Sarpong on the development of synthetic strategies to various fused N-heterocycles. He began his independent career at North Carolina State University in 2016, working on the development of novel synthetic methods involving strained ring systems. |

|
Yu-Che Chang was born and raised in Taiwan. He received his B.S. and M.S. from National Tsing Hua University in 2017 and 2019. He is currently a fourth-year graduate student in Prof. M. Kevin Brown's laboratory at Indiana University, Bloomington. His research mainly focuses on photochemical strain-release cycloadditions. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved