Org. Synth. 2025, 102, 217-235
DOI: 10.15227/orgsyn.102.0217
Large-Scale Syntheses of Silanoxy-Tetrahydrofurans via Stereospecific [5,5]-Rearrangements of Silanol Epoxides
Submitted by Silver Raju
1 and Shyam Sathyamoorthi*
,1Checked by Nnamdi Akporji and Kevin Campos
1. Procedure (Note 1)
A. Preparation of (E)-di-tert-butyl(hex-4-en-1-yloxy)silanol (1). A single-neck (24/40 joint) 500-mL round-bottom flask is equipped with a Teflon-coated magnetic stir bar (2.5 cm x 0.5 cm, oval-shaped). The vessel is purged with nitrogen from a Schlenk line connected to an oil bubbler using a 20G inlet and a 20G exit needle for 30 min and dried with a heat gun (Figure 1A). The flask is then charged with di-tert-butylsilyl bis(trifluoromethanesulfonate) (21.4 mL, 28.9 g, 65.6 mmol, 1.5 equiv) (Note 2) using a plastic syringe fitted with a 22G needle and CH2Cl2 (174 mL) (Note 3) using a plastic syringe fitted with a 20G needle (Figure 1B). The reaction flask is then placed in an ice-water bath maintained at 0 ℃. After ten minutes, 2,6-lutidine (14.9 mL, 13.7 g, 128 mmol, 3 equiv) (Note 4) is then added dropwise via syringe pump (KDS LegatoTM 101, dual syringe nanoliter pump) and a plastic syringe fitted with a 20G needle (total time of addition = 10 min). Subsequently, the reaction mixture is stirred (500 rpm) for 15 min at 0 ℃. trans-4-Hexen-1-ol (98 wt%, 5.22 mL, 4.44 g, 43.9 mmol, 1 equiv) (Note 5) is then added dropwise via syringe pump and a plastic syringe fitted with a 20G needle over 10 minutes. Stirring (500 rpm) is continued at 0 ℃ for 1 hour (Figure 1C).

Figure 1. Reaction set-up for Step 1. A. Reaction flask purging under nitrogen; B. Reaction flask with di-tert-butylsilyl bis(trifluoro-methanesulfonate) and CH2Cl2 at ambient temperature; C. After cooling to 0 ℃ and after addition of 2,6-lutidine and trans-4-hexen-1-ol
The progress of the reaction is monitored by thin-layer chromatography (TLC) (Figure 2A) (Note 6), and starting material (trans-4-hexen-1-ol) is consumed in approximately 1 h following addition. Then, the reaction is quenched at 0 ℃ by careful, portion-wise addition of saturated aqueous NaHCO3 solution (75 mL). The reaction mixture is then transferred to a 1-L separatory funnel (Figure 2B). The aqueous layer is drained, and the organic layer is washed with additional saturated, aqueous NaHCO3 solution (3 portions of 75 mL each). The organic layer is then washed with 1 M aqueous HCl solution (2 portions of 75 mL each) (Note 7). The combined organic layers are dried over MgSO4 (50 g) (Note 8), are filtered through a CHEMRUS 60-mL disposable plastic filter funnel (CR-1018-40 with a medium porosity frit in place) into a 1-L round-bottom flask and concentrated under reduced pressure via a rotary evaporator.
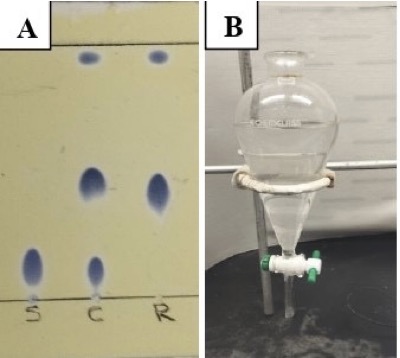
Figure 2. A. The progress of the reaction is monitored by TLC analysis on silica gel with 1:1 hexanes/CH2Cl2 as the eluent. Spots are visualized after the plate is dipped in Ceric Ammonium Molybdate solution (Note 6) and heated. The starting material, trans-4-hexen-1-ol has an Rf of 0.17 (bottom spot) and silanol product 1 has an Rf of 0.48 (middle spot). The top spot is a dimeric side-product. Note: S = starting material, C = co-spot lane, P = product B. The separation of layers in the reaction workup
The resulting residue (Figure 3A) is dissolved in toluene (10 mL) (Note 9), and the solids are filtered through a filter funnel (CHEMRUS 60mL disposable plastic filter funnel (CR-1018-40) with a medium porosity frit in place). The crude mixture is purified by flash chromatography utilizing a Teledyne Combiflash ISCO System on silica gel (Figure 3B and Notes 10, 11). The fractions containing the product (Figure 3B) were combined and concentrated under reduced pressure on a rotary evaporator to give 1 as a colorless oil (7.4 g, 28.6 mmol, 65% yield, Note 12) (Figure 3C). The purity of 1 was determined to be 95.1 wt% by qNMR using 1,3,5-trimethoxybenzene as the internal standard (Note 13).
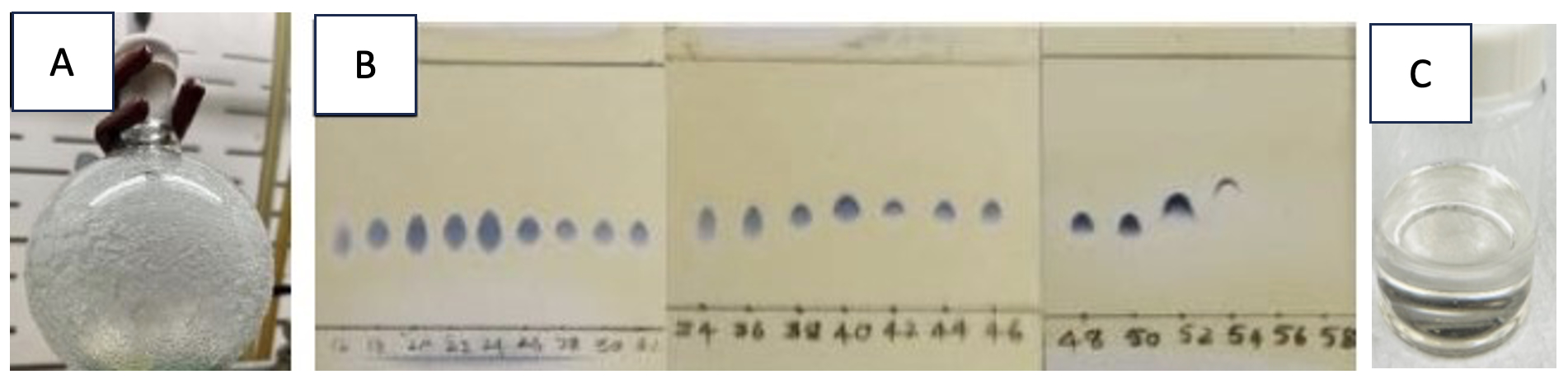
Figure 3. A. Product before purification and filtration. The eluent used during purification by chromatography was 95:5 hexanes/CH2Cl2 (500 mL), 90:10 hexanes/CH2Cl2 (500 mL), 85:15 hexanes/CH2Cl2 (2000 mL). Fraction sizes of 20 mL were collected. B. Fractions were checked using thin-layer chromatography (1:1 hexanes/dichloromethane) and visualized by heating with ceric ammonium molybdate stain. C. (E)-di-tert-butyl(hex-4-en-1-yloxy)silanol is a colorless oil
B. Preparation of di-tert-butyl(3-((2R*,3R*)-3-methyloxiran-2-yl)propoxy)silanol (2). A single-neck (24/40 joint) 500-mL round-bottom flask is equipped with a Teflon-coated magnetic stir bar (2 cm x 0.5 cm, oval-shaped). The vessel is purged with nitrogen from a Schlenk line connected to an oil bubbler using a 20G inlet and a 20G exit needle for 30 min (Figure 4A). The flask is then charged with (E)-di-tert-butyl(hex-4-en-1-yloxy) silanol 1 (5.8 g, 22 mmol, 1 equiv) using a plastic syringe fitted with a 22G needle and CH2Cl2 (224 mL) using a plastic syringe fitted with a 20G needle. The reaction flask is then placed in an ice-water bath maintained at 0 ℃. m-Chloroperoxybenzoic acid (7.7 g, 45 mmol, 2 equiv) (Note 14) is added portion-wise over 10 min. The reaction flask is capped with a rubber septum fitted with a nitrogen Schlenk line using a 20G inlet. The reaction mixture is stirred (600 rpm) for 5 min at 0 ℃, and the ice-water bath is then removed. The reaction mixture is allowed to warm to room temperature with stirring over a period of 2 h (Figure 4B-D). The progress of the reaction is monitored by silica gel TLC using 10% EtOAc in CH2Cl2 as the eluent, and spots are visualized by staining with ceric ammonium molybdate solution. The starting material 1 has an Rf of 0.80 and epoxide product 2 has an Rf of 0.40. After full consumption of the starting material (~2 h), the reaction is cooled to 0 ℃ using an ice-water bath and quenched by slowly adding saturated aqueous sodium thiosulfate solution (50 mL) (Note 15).

Figure 4. A. Reaction flask purging under nitrogen; B. Compound 1 dissolved in CH2Cl2 and reaction mixture cooled to 0 ℃ using an ice-water bath prior to addition of mCPBA; C. Directly after addition of mCPBA; D. After warming to room temperature, close to reaction completion
The reaction mixture is then transferred to a 1-L separatory funnel containing 75 mL of MTBE (Note 16). The aqueous layer is removed, and the organic layer is washed with 1 M aqueous NaOH solution (3 portions of 75 mL each) (Note 17). The organic layer is then collected, dried over MgSO4 (50 g), filtered through a CHEMRUS 60-mL disposable plastic filter funnel (CR-1018-40 with a medium porosity frit in place) into a 1-L round-bottom flask, and concentrated under reduced pressure using a rotary evaporator. The resulting residue is dissolved in CH2Cl2 and purified by flash chromatography utilizing a Teledyne Combiflash ISCO System on silica gel (Note 18). The fractions containing product were combined and concentrated under reduced pressure on a rotary evaporator to afford 2 (Figure 5A) as a colorless oil (5.0 g, 18 mmol, 81% yield, Figure 5B, Note 19). The purity of 2 (Note 20) was determined to be 98.4 wt% by qNMR using 1,3,5-trimethoxybenzene as the internal standard.
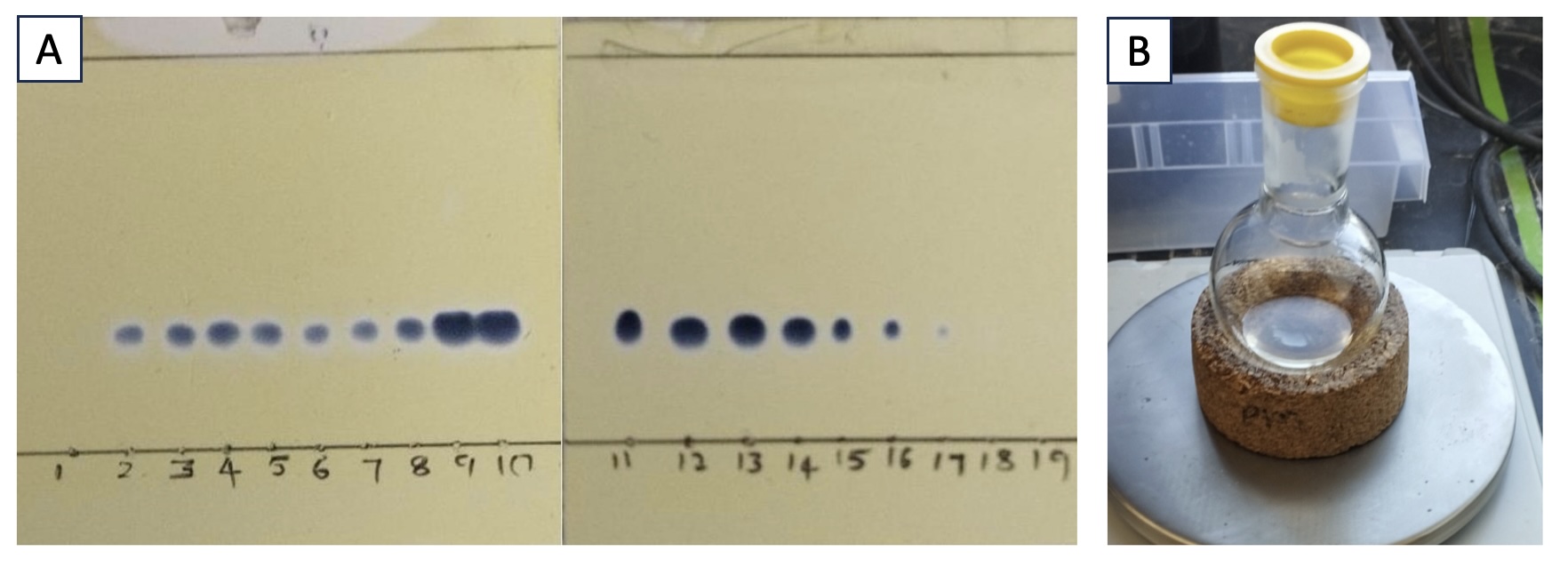
Figure 5. A. Fractions were analyzed for product using thin-layer chromatography plates (eluted with 10% EtOAc/CH2Cl2) stained with ceric ammonium molybdate; B. Pure di-tert-butyl(((2R*,3R*)-3-methyloxiran-2-yl)propoxy)silanol (Compound 2) is a colorless oil
C. Preparation of di-tert-butyl((R*)-1-((S*)-tetrahydrofuran-2-yl)ethoxy) silanol (3). A single-neck (24/40 joint) 500-mL round-bottom flask is equipped with a Teflon-coated magnetic stir bar (2.5 cm x 0.5 cm, oval-shaped). The vessel is purged with nitrogen from a Schlenk line connected to an oil bubbler using a 20G inlet and a 20G exit needle for 30 min (Figure 6). The flask is then charged with di-tert-butyl(3-((2R*,3R*)-3-methyloxiran-2-yl)propoxy) silanol 2 (5.0 g, 18.2 mmol, 1 equiv) using a plastic syringe fitted with a 22G needle and CH2Cl2 (172 mL) using a plastic syringe fitted with 20G needle (Figure 6A). NaHCO3 (1.53 g, 18.2 mmol, 1 equiv) (Note 21) is added in one portion. The reaction flask is then placed in an ice-water bath maintained at 0 ℃ and stirring (500 rpm) is commenced (Figure 6C). After 10 min, Ph3CBF4 (902 mg, 2.73 mmol, 0.15 equiv) (Note 22) is added portion-wise over 5 min, and the reaction flask is capped with a rubber septum fitted with a nitrogen Schlenk line using a 20G needle. The ice-water bath is removed, and the reaction mixture is warmed to room temperature over the next 2 h (Figure 6D).

Figure 6. A. Start of the reaction with Compound 2 dissolved in CH2Cl2; B. After addition of NaHCO3; C. The reaction is cooled to 0 ℃ prior to addition of Ph3CBF4; D. After addition of Ph3CBF4 (at 0 ℃), the reaction mixture turns bright yellow, and this color persists even after warming to room temperature.
The progress of the reaction is monitored by TLC (Figure 7A). The starting material 2 has an Rf of 0.25, the product 3 has an Rf of 0.50, and the catalyst impurity has an Rf of 0.85 (Note 23). After consumption of starting material (~2 h), the reaction mixture is cooled to 0 ℃ using an ice-water bath, quenched by adding saturated aqueous NaHCO3 solution (50 mL), and further diluted with an additional 20 mL of CH2Cl2. The reaction mixture is transferred to a 1-L separatory funnel. The aqueous layer is removed, and the organic layer is washed with saturated aqueous NaHCO3 solution (2 portions of 50 mL each). The pale-yellow organic layer is collected, dried over MgSO4 (50 g), and filtered through a CHEMRUS 60-mL disposable plastic filter funnel (CR-1018-40 with a medium porosity frit in place) into a 1-L round bottom flask. After concentrating under reduced pressure on a rotary evaporator, the resulting residue is dissolved in 10 mL of CH2Cl2 and purified by flash chromatography utilizing a Teledyne Combiflash ISCO System on silica gel using 0-40% EtOAc/CH2Cl2 as the solvent system (Figure 7B). The fractions containing product are combined and concentrated under reduced pressure on a rotary evaporator to give 3 as a colorless oil (Figure 7C) (3.15 g, 11.5 mmol, 63% yield, Note 24) with 98.9 wt% purity (Note 25).
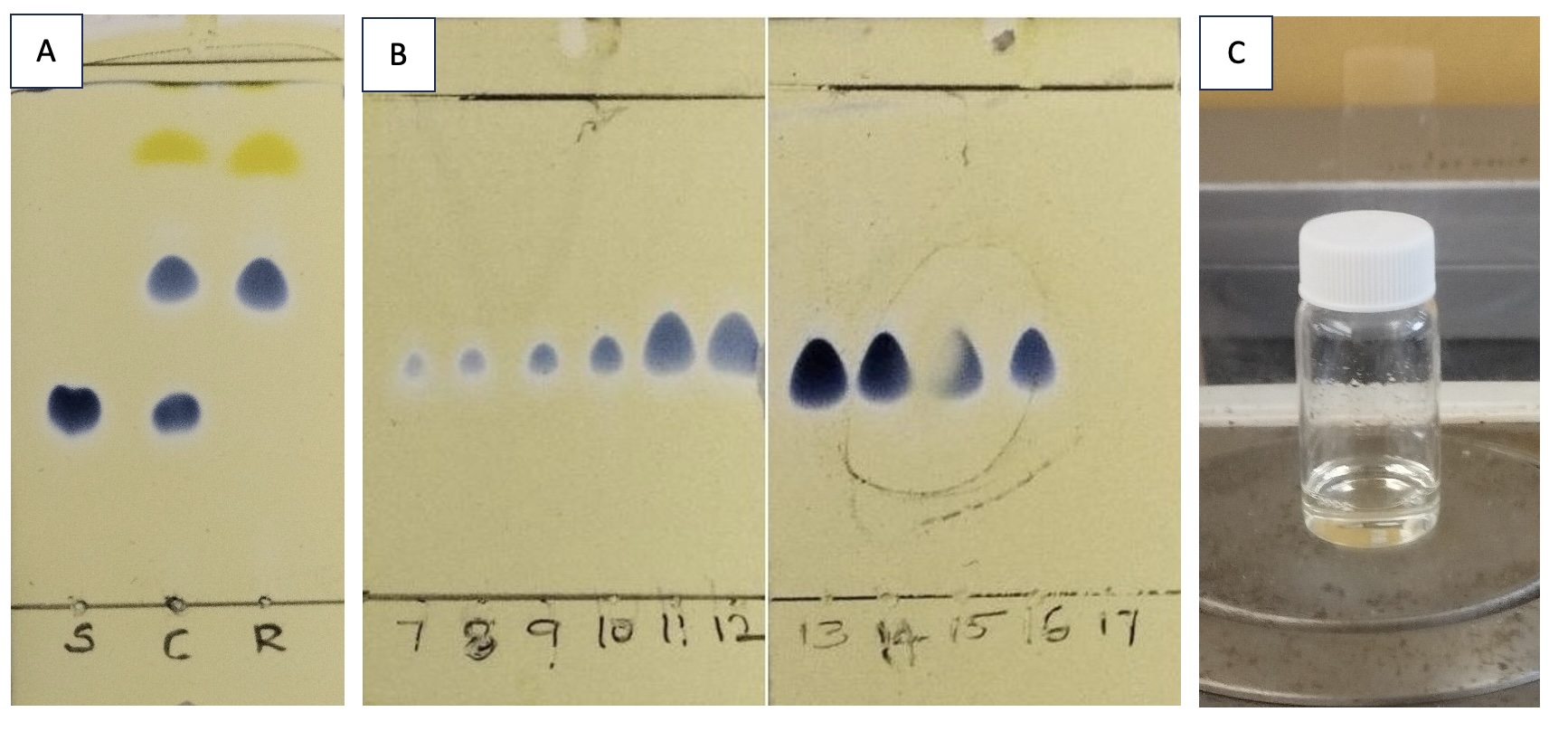
Figure 7. A. The progress of the reaction is monitored by TLC analysis on silica gel with 5% EtOAc in CH2Cl2 as the eluent. Spots are visualized after the plate is dipped in ceric ammonium molybdate stain and heated; B. In a similar fashion, chromatographic fractions were checked for product using thin-layer chromatography plates; C. Di-tert-butyl((R*)-1-((S*)-tetrahydrofuran-2-yl)ethoxy)silanol 3 is a colorless oil after purification.
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/about/governance/committees/chemical-safety.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
di-tert-butylsilyl bis(trifluoromethanesulfonate),
m-chloroperoxybenzoic acid,
diethyl ether,
acetone,
toluene,
CH2Cl2,
trans-4-hexen-1-ol,
2,6-lutidine,
concentrated hydrochloric acid,
magnesium sulfate, silica gel, hexanes,
acetone, concentrated
sulfuric acid,
sodium bicarbonate,
sodium thiosulfate pentahydrate,
sodium hydroxide,
ammonium molybdate, and
ceric ammonium molybdate.
2.
Di-tert-butylsilyl bis(trifluoromethanesulfonate) was purchased from Combi-Blocks (purity of 96%) and used as received.
3.
CH2Cl2 was purchased from Sigma-Aldrich (≥99.5%, ACS reagent grade) and used as received.
4.
2,6-Lutidine was purchased from TCI America (purity > 98%) and used as received.
5.
trans-4-Hexen-1-ol was purchased from Ambeed (purity 98%) and used as received.
6.
Ceric ammonium molybdate stain was prepared by dissolving 12 g of
ammonium molybdate, 0.5 g of
ceric ammonium molybdate, and 15 mL of concentrated
H2SO4 in 235 mL water. TLC plates was briefly submerged into the stain and heated until spots were fully developed.
7.
Hydrochloric acid (Certified ACS Plus, 36.5 to 38%) was purchased from Fisher Chemical. 1 M
HCl solution was prepared by adding 83 mL of this to 917 mL of deionized water.
8.
Magnesium sulfate (anhydrous) was purchased from Fisher Scientific and used directly. The checker used a filter funnel to remove
magnesium sulfate, followed by a rinse of the cake with 10 mL of
DCM (CHEMRUS 60mL disposable plastic filter funnel (CR-1018-40) with a medium porosity frit).
9.
Toluene was purchased from Sigma-Aldrich (≥99.5%, ACS reagent grade) and used as received.
10. Silica Gel (Grade 60, 230-400 mesh) was purchased from Fisher Scientific.
11. Hexanes (>98.5%) was purchased from Fisher Scientific and used as received.
12.
(E)-di-tert-butyl(hex-4-en-1-yloxy)silanol 1 characterization:
1H NMR
pdf (500 MHz, Chloroform-
d) δ 5.49 - 5.39 (m, 2H), 3.78 (t,
J = 6.4 Hz, 2H), 2.09 - 2.02 (m, 2H), 1.66 - 1.63 (m, 3H), 1.62 - 1.58 (m, 2H), 1.02 (s, 18H).
13C NMR
pdf (126 MHz, CDCl
3) δ 131.2, 125.2, 63.0, 32.9, 29.0, 27.6, 20.6, 18.1. IR ν 3501, 2934, 2860, 1471, 1106, 827 cm
-1. HRMS (ESI) m/z = [M + Na]
+ calculated for C
14H
30O
2SiNa
+ 281.1913. Found 281.1909.
13. The checkers observed in the first run, the product was a pale-yellow oil, however the purity was still high.
pdf14.
m-Chloroperoxybenzoic acid (70-75% pure, with a balance of
3-chlorobenzoic acid and water) was purchased from Thermo Scientific and was used without purification.
15.
Sodium thiosulfate pentahydrate (Fisher chemical, purity >99%) was purchased from Fisher Scientific and used as is.
16. The checker used MTBE purchased from Sigma Aldrich in the place of
Et2O used by the authors.
17.
Sodium hydroxide pellets were purchased from Acros Organics. 1 M
NaOH solution was prepared by dissolving 20 g of
NaOH pellets in 500 mL deionized water.
18. The checker used 0-30%
EtOAc/
DCM as the mobile phase in column chromatography utilizing a Teledyne Combiflash ISCO System.
19.
di-tert-Butyl (3-((2R*,3R*)-3-methyloxiran-2-yl)propoxy)silanol 2 characterization:
1H NMR
pdf (500 MHz, chloroform-
d) δ 3.91 - 3.82 (m, 2H), 2.81 - 2.63 (m, 2H), 2.59 (s, 1H), 1.79 - 1.62 (m, 3H), 1.56 (m, 1H), 1.30 (d,
J = 5.2 Hz, 3H), 1.02 (s, 9H), 1.00 (s, 9H).
13C NMR
pdf (126 MHz, CDCl
3) δ 62.7, 60.2, 54.6, 29.8, 28.2, 27.7, 27.6, 20.8, 20.6, 17.7. IR ν 3437, 2933, 2860, 1473, 1130, 650 cm
-1. HRMS (ESI) m/z = [M+Na]
+ calculated for C
14H
30O
3SiNa
+ 297.1862. Found 297.1871.
20. The purity of
2 was determined to be 98.4 wt% by qNMR
pdf using
1,3,5-trimethoxybenzene as the internal standard.
21.
Sodium bicarbonate (ACS reagent grade) was purchased from Oakwood Chemical and used as received.
22.
Triphenylcarbenium tetrafluoroborate (
Ph3CBF4) was purchased from Alfa Aesar (reported purity of 97%) and used as received.
23. The checker used 5%
EtOAc/
DCM as the mobile phase for TLC (R
f: 0.50).
24.
Di-tert-Butyl((R*)-1-((S*)-tetrahydrofuran-2-yl)ethoxy)silanol (
3) characterization
1H NMR
pdf (500 MHz, chloroform-
d) δ 4.43-4.38 (m, 1H), 3.97 - 3.89 (m, 1H), 3.76 - 3.72 (m, 1H), 3.67-3.62 (m, 1H), 1.96-1.87 (m, 3H), 1.79 - 1.72 (m, 1H), 1.16-1.12 (m, 3H), 1.04 (s, 9H), 0.99 (s, 9H).
13C NMR
pdf (126 MHz, CDCl
3) δ 84.2, 68.1, 67.7, 27.9, 27.7, 25.2, 23.3, 21.2, 20.5, 20.3. IR ν 3436, 2963, 2860, 1475, 650 cm
-1. HRMS (ESI) m/z = [M + Na]
+ calculated for C
14H
30O
3SiNa
+ 297.1862. Found 297.1881.
25. The checker used 0-40%
EtOAc/
DCM as the mobile phase in column chromatography utilizing a Teledyne Combiflash ISCO System. The purity and recovery were high.
pdf
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Our laboratory
1 is dedicated to transforming the di-
tert-butyl silanol functional group into a synthetically valuable auxiliary.
2,3,4,5,6,7,8,9 During our investigation into epoxide opening reactions facilitated by pendant silanols, we serendipitously discovered an unprecedented rearrangement of silanol epoxides into 1'-silanoxy-tetrahydrofurans.
10 The uniqueness of this transformation encouraged us to further explore its scope, mechanism, and potential applications.
Our study, detailed in this Org. Synth. contribution, presents a broadly applicable rearrangement reaction for various trans-disubstituted and cis-disubstituted epoxides, as well as heteroatom-containing epoxides (Figures 8, 9, and 10). This reaction efficiently yields a corresponding array of 1'-silanoxy-tetrahydrofurans and 1'-silanoxy-tetrahydropyrans. Notably, we successfully applied this method in the concise syntheses of (±)-solerone and (±)-muricatacin (Figure 11).
Figure 8. Substrate Scope (trans epoxides)
Figure 9. Substrate Scope (cis epoxides)
Figure 10. Preparation of Silanoxy-Tetrahydropyrans
Figure 11. Application of the rearrangement reaction for the syntheses of (A) (±)-solerone and (B) (±)-muricatacin
For the syntheses of silanoxy-tetrahydrofurans, we term these [5,5]-rearrangements, for the ring sizes involved in epoxide opening and silyl transfer (Figure 12). By analogy, for the syntheses of silanoxy-tetrahydropyrans, [6,5]-rearrangements occur.
Figure 12. Proposed mechanism for these rearrangement reactions
With TBDPS substrate 78, rearrangement occurred in a lower yield (Figure 13A). Thus, the distal oxygen of the silanol was not required for a productive reaction, but its presence did promote product formation. With silanoxy methyl ether substrate 80, the yield of rearranged product 81 was higher. Both substrates were outperformed by silanol 1 (Figure 8, Substrate 1 and Product 2), which rearranged in a 78% yield. We found that one-pot epoxidation-cyclization reactions of alkenyl alcohols were possible using mCPBA, but the mass balance and product yields of these reactions were generally poor (Figure 13B). Thus, we recommend protecting the alcohol and then implementing a two-step protocol of epoxidation and rearrangement.
Figure 13. Use of the silanol auxiliary does provide synthetic advantages
Appendix
Chemical Abstracts Nomenclature (Registry Number)
Di-tert-butylsilyl bis(trifluoromethanesulfonate); (85272-31-7)
2,6-Lutidine; (108-48-5)
Meta-Chloroperoxybenzoic acid; (937-14-4)
Triphenylcarbenium tetrafluoroborate; (341-02-6)
Sodium Bicarbonate; (144-55-8)

|
Silver Raju received his B.Sc. from Kakathiya University and his M.Sc. in Organic Chemistry in 2015 from Osmania University, India. He completed his Ph.D. in Organic Chemistry in 2024 at the CSIR-Indian Institute of Chemical Technology in Hyderabad, under the supervision of Dr. Srivari Chandrasekhar. Subsequently, he joined the Department of Medicinal Chemistry at the University of Kansas, Lawrence, KS, USA, as a Postdoctoral Research Fellow in Prof. Shyam Sathyamoorthi's group. |

|
Shyam Sathyamoorthi completed a B.S. degree in Cell and Molecular Biology with a minor in Chemistry at Tulane University, New Orleans, Louisiana, where he worked in the labs of Professor Ken Muneoka and Professor Robert A. Pascal, Jr. He then completed a PhD in chemistry at Stanford University under the guidance of Professor Richard N. Zare (2018) as well as a Doctor of Medicine degree at the Stanford University School of Medicine (2019). In July 2019, he started his independent career as an assistant professor in the Department of Medicinal Chemistry at the University of Kansas, Lawrence, KS, USA. He was promoted to associate professor (with tenure) in 2024. |

|
Nnamdi Akporji is originally from Lagos, Nigeria and grew up in the Washington DC metro area. He obtained his B.S. in chemistry at the Rochester Institute of Technology working with Dr. Hans Schmitthenner. Later, he obtained his PhD in organic chemistry in 2021 at the University of California Santa Barbara under the tutelage of Dr. Bruce Lipshutz. He is currently working at Merck Research Laboratories based in Rahway NJ in the process chemistry department. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved