Org. Synth. 2025, 102, 335-349
DOI: 10.15227/orgsyn.102.0335
Synthesis of 6-Methoxy-2-methyl-2-(4-methylpent-3-en-1-yl)chroman-4-one via Kabbe Condensation
Submitted by Stephen A. Chamness
✝𐊔1, Emily F. Traficante
✝𐊔1, Trenton R. Vogel
✝𐊔1, and Corinna S. Schindler*
‡𐊔1Checked by Max Neumann and Tehshik Yoon
1. Procedure (Note 1)
2-Methyl-2-(4-methylpent-3-en-1-yl)chroman-4-one (3). The apparatus is flame-dried under vacuum and maintained under an atmosphere of nitrogen during the course of the reaction. A 100-mL, 14/20 single-necked, round-bottom flask equipped with a 2-cm Teflon-coated, magnetic egg-shaped stir bar, and a rubber septum with a pressure-equalizing nitrogen needle inlet is charged with freshly distilled pyrrolidine (1.48 mL, 18.1 mmol, 1.50 equiv) (Note 2) and anhydrous DMSO (19.3 mL) (Note 3). Then, butyric acid (0.55 mL, 6.02 mmol, 0.50 equiv) (Note 4) is added via syringe to the solution over thirty seconds. A white gas forms in the headspace of the flask while the butyric acid is added (Figure 1). Once added, the acid/base mixture is stirred for 10 min.
Figure 1. Butyric acid addition to the pyrrolidine solution and gas production over time. A. Start of butyric acid addition; B. Part-way through the addition; C. Completion of butyric acid addition with significant gas evolution visible.
Then, 6-methyl-5-hepten-2-one (2) (1.96 mL, 13.2 mmol, 1.10 equiv) (Note 5) is added to the mixture via syringe over thirty seconds and the mixture is stirred for 15 min. As the solution stirs, the color changes from colorless to pale yellow. 2'-Hydroxy-5'-methoxyacetophenone (1) (2.00 g, 12.0 mmol, 1.00 equiv) (Note 6) is added in one portion as a solution in DMSO (3.8 mL), then the scintillation vial containing the acetophenone is rinsed with DMSO (1 mL) and the solution is also added to the reaction flask. Upon addition of the acetophenone to the reaction, an immediate color change is observed from pale yellow, to orange, to red/orange (Figure 2). The resulting mixture is stirred for 18 h at ambient temperature and monitored by TLC (Note 7).

Figure 2. Acetophenone addition overtime and corresponding color change of the reaction solution. A. Start of acetophenone addition; B. Solution after some added; C. Most of the acetophenone added; D. Solution upon completion of acetophenone addition; E. 18 h after acetophenone addition.
The reaction mixture is then transferred to a 2-L separatory funnel and is diluted with deionized water (1200 mL, 100 mL/mmol) (Note 8) and ethyl acetate (600 mL, 50 mL/mmol) (Note 9) (Figure 3). The organic layer is separated (Note 10), and the aqueous layer is extracted with ethyl acetate (2 x 300 mL) (25 mL/mmol). The organic layers are combined and washed with brine (600 mL, 50 mL/mmol) (Note 11). The combined organic layer is then dried via addition of 5 g of MgSO4 (Note 12) which is allowed to sit for 5 min. Following this, the organic layer is gravity-filtered through a Qualitative P5 filter paper with medium porosity at a slow flow rate and concentrated at 40 ℃ by rotary evaporation (200 mbar) and then at 0.1 mbar to provide the product as a crude reside. At this point, the crude material appears as a reddish-brown oil.

Figure 3. Aqueous workup of reaction in separatory funnel with ethyl acetate. A. Post-mixing of crude reaction solution added to water and ethyl acetate; B. Separation of aqueous (bottom) and organic (top, brown) layers; C. Brine wash of organic layers separate into aqueous (bottom) and organic (top, yellow).
A column (50 mm diameter) is slurry packed to a height of 8 in. of silica gel (Notes 13-14) using hexanes and topped with 1 in. of sand (Figure 4) (Note 15), and the product is charged onto the column. Fraction collection (20 mL fractions) is begun, and the column is eluted with 1500 - 2000 mL of 5% ethyl acetate-hexanes (Note 16) (19:1 hexanes : ethyl acetate). The desired product elutes between fractions 40-65, which are concentrated by rotary evaporation (40 ℃, 150 mm Hg) (Notes 17-18). Following rotary evaporation, the organic residue is further dried under high vacuum (0.1 mm Hg) for at least 2 h to yield pure 2-methyl-2-(4-methylpent-3-en-1-yl)chroman-4-one (3) (2.46 g, 75% yield, >98 % purity by quantitative NMR) as a transparent yellow oil (Figure 5) (Notes 19,20,21,22).

Figure 4: Column chromatography of reaction on silica gel utilizing an eluent system of 19:1 Hexanes: Ethyl acetate. A. Concentrated crude reaction mixture prior to column loading; B. Column after loading with crude material; C. Column in progress and start of band separation; D. Column upon completion; The yellow band at the bottom is starting material.
Figure 5: Purified 2-methyl-2-(4-methylpent-3-en-1-yl)chroman-4-one (3), appearing as a yellow oil.
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/about/governance/committees/chemical-safety.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
2'-hydroxy-5'-methoxyacetophenone,
6-methyl-5-hepten-2-one,
butyric acid,
pyrrolidine,
dimethyl sulfoxide (
DMSO),
ethyl acetate (
EtOAc),
hexane,
magnesium sulfate (
MgSO4),
sodium hydroxide (
NaOH),
sodium chloride (
NaCl), and silica, as well as the proper procedures for Hickman distillation for the purification of
pyrrolidine.2.
Pyrrolidine (99 %) was purchased from Thermo Scientific (A14852.AP) and was purified via Hickman distillation (Figure 6). The Hickman still was oven dried at 150 ℃ and cooled under a stream of nitrogen with an outlet needle prior to use. The outlet needle was removed, and
pyrrolidine was charged into the bottom of the distillation apparatus using a straightened long oven-dried needle attached to a syringe to reach the bottom bulb of the Hickman still. A Master heat gun (HG.301A, 149/260 ℃) was utilized to heat the
pyrrolidine at the highest temperature setting and the distilled
pyrrolidine was collected. To prevent bumping and overheating the glass, it is advised to slowly move the heat gun around the bottom of the bulb constantly while performing the distillation. It is also best to use distilled
pyrrolidine for this transformation as unpurified
pyrrolidine has been found to give inconsistent reaction yields. The checkers note that
pyrrolidine can also be purified by short-path distillation (14/20 joints, 110 mm width x 115 mm height) at atmospheric pressure (heated by oil bath set to 100 ℃).

Figure 6. Hickman distillation of pyrrolidine. A) Cooling oven-dried Hickman still under N2; B) Initial stages of distillation with heat gun; C). Collection of pure pyrrolidine observed in collection vessel; D) Completion of distillation.
3.
Dimethyl sulfoxide (
DMSO) (99.9 %) was purchased from Fisher Scientific (AA36480M6) and was freshly dispensed from the B.C. Enterprises solvent purification system. The checkers note that purification of
DMSO by a solvent purification system is not required.
DMSO was stored over activated 4 Å molecular sieves (1 g/5 mL) for 18 h and degassed by sparging with nitrogen for 20 min using a long inlet needle submerged in the solvent, while a second needle pierced the septum to allow for continuous sparging of the solvent. Other solvents were analyzed for the transformation including
N,N′-dimethylimidazolidinone (DMI),
N,N-Dimethylacetamide (DMA),
Sulfolane, and
Acetonitrile. DMI (>99 %) that was purchased from Fisher Scientific (D1477100ML) and used without further purification gave comparable results; however, a fresh bottle of DMI must be used as the yield decreased for old, opened bottles of DMI.
4.
Butyric acid (>99 %) was purchased from Acros (108111000) and used without further purification.
5.
6-Methyl-5-hepten-2-one (99 %) was purchased from Sigma-Aldrich (M48805-100ML) and used without further purification.
6.
2'-Hydroxy-5'-methoxyacetophenone (99 %) was purchased from Sigma-Aldrich (114995-25G) and used without further purification.
7. Thin layer chromatography (TLC) was conducted on Merck silica gel 60 F254 plates. The eluent system was 1:9
ethyl acetate:hexanes (Figure 7A).The plate was air dried then visualized with both long wave (365 nm) and short wave (254 nm) UV light. Under short wave UV, both the product (Rf = 0.43) and starting material (Rf = 0.31) are visible (Figure 7B). Under long wave UV, the product appears strongly whereas the starting material is not apparent (Figure 7C).
Figure 7: Crude reaction TLCs run in 9:1 Hexanes: Ethyl acetate on glass-baked silica plates. A. TLC showing Rf values of the SM, Co, and Rxn; B. TLC under short wave UV, 254 nm. C. TLC under long wave UV, 365 nm. (SM = acetophenone 1, Co = Co-spot of SM and Rxn, Rxn = crude reaction mixture)
8. In-house deionized water was used as received.
9.
Ethyl acetate (>99.5 %) was purchased from Fisher Scientific (E145-20) and was used without further purification.
10. The organic layer takes some time to fully separate from the aqueous layer. The mixture appears an opaque yellow and as it separates, the organic layer becomes brown. Subsequent washes of the aqueous layer will produce an organic layer that is more yellow.
11. Saturated
sodium chloride solution was made from adding
sodium chloride, (>99.0 %) purchased from Sigma-Aldrich (S9888-10KG) and used without further purification., to in-house deionized water until solvation no longer occurs.
12.
Magnesium sulfate (>99.5 %) was purchased from Sigma-Aldrich (M7506-12KG) and was used without further purification.
13. SiliaFlash irregular silica gel P60, 40 - 63 μm, 60 Å was purchased from SiliCycle (R12030B-25KG) and was used without further purification.
14. The column dimensions were chosen and modified based on
https://labs.chem.ucsb.edu/zakarian/armen/how-to-do-flash-column-3.pdf. The bottom of the column was packed with cotton, and then a layer of sand (~1 in) was added. A slurry of silica in hexanes was then added to the column to result in the desired height of silica after packing. The crude material was wet loaded onto the column, rinsing the crude reaction container thrice with hexanes. Product elutes soon after an impurity that is yellow/green in color. As the product comes off the column, it appears colorless in color, and as the column nears completion, remaining starting material begins to co-elute with the product. The starting material comes off as a very bright yellow.
15. Sand (Sea) was purchased from Sigma-Aldrich (SX0076-1) and was used without further purification.
16. Hexanes (>98.5 %) was purchased from Fisher Scientific (H292-20) and was used without further purification.
17. An impurity faintly visible on TLC via staining with
potassium permanganate (
KMnO4) is sometimes observed immediately below the product spot, and care should be taken when collecting fractions to test TLC by stain in addition to UV light to ensure the highest purity.
18. If the reaction does not go to completion and residual starting material remains in the crude reaction mixture (as determined by a characteristic phenol peak in
1H NMR near 12 ppm), the mixture can be diluted in
ethyl acetate and washed with 15 wt % aq
NaOH (3 - 5 times) to effectively remove the remaining starting material.
19. Characterization data for product
3:
1H NMR
pdf (500 MHz, CDCl
3) δ 7.29 (d,
J = 3.2 Hz, 1H), 7.08 (dd,
J = 9.0, 3.2 Hz, 1H), 6.86 (d,
J = 9.0 Hz, 1H), 5.06 (t,
J = 7.1 Hz, 1H), 3.80 (s, 3H), 2.76 (d,
J = 16.5 Hz, 1H), 2.65 (d,
J = 16.5 Hz, 1H), 2.16 - 2.03 (m, 2H), 1.85 - 1.75 (m, 1H), 1.71 - 1.62 (m, 4H), 1.57 (d,
J = 1.4 Hz, 3H), 1.40 (s, 3H).
13C NMR
pdf (126 MHz, CDCl
3) δ 192.7, 154.5, 153.6, 132.3, 125.3, 123.4, 120.1, 119.6, 107.0, 80.9, 55.8, 47.5, 39.1, 25.6, 23.9, 22.3, 17.6. IR
pdf (neat liquid) ν
max: 2970, 2915, 1685, 1618, 1484, 1429, 1281, 1217, 1077, 1034, 826, 802, 701. bp = 110 ℃ at 15 mm Hg HRMS (ESI+)
m/z calc'd. for C
17H
23O
3+ [M+H
+]: 275.1642; Found: 275.1642.
20. The checkers reported 1.25 g (76 %) on half scale and 2.46 g (75 %) on full scale with purities of >98 %.
21. Product purity was determined by quantitative
1H NMR
pdf (with a 30 s relaxation delay between scans for a 16-scan experiment) using
dimethyl terephthalate as the internal standard. The purity was calculated to be 99 wt % taking the integral of the product
3 signal at 3.79 ppm (s, 3H) was compared with the integral of the
dimethyl terephthalate peak at 3.94 ppm (s, 3 H).
22.
Dimethyl terephthalate (>99 %) was purchased from Sigma-Aldrich (185124-500G) and was used without further purification.
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
The Kabbe condensation entails the combination of an aliphatic ketone with either a salicylaldehyde or 2'-hydroxyacetophenone to form bicyclic chromanes or chromanones
2,3,4. These bicyclic scaffolds are sought-after as privileged structures given their frequent occurrence in plant-derived natural products and for their use as pharmaceutical agents
5,6,7. In early development, this reaction was performed using enamines derived from the corresponding ketones as reaction partners with 2'-hydroxyacetophenone derivatives to form chroman-4-ones. Later modifications demonstrated the reaction can progress with
in-situ generation of an enamine-iminium equilibrium from the combination of the corresponding aliphatic ketone and amine precursors, simplifying the synthetic protocol. However, this transformation still typically required harsh conditions to favor the forward reaction (i.e., reflux in toluene with a Dean-Stark apparatus)
8. More recently, Bapodra and co-workers disclosed a mild organocatalyzed Kabbe condensation protocol to synthesize 2,2-dialkylchroman-4-ones
9. This reaction operates at ambient temperature and tolerates several aryl substituents on the 2'-hydroxyacetophenone partners, in addition to multiple cyclic and acyclic alkyl ketones. Table 1 demonstrates the scope of this organocatalyzed procedure. Our lab recently reported a modified version of this procedure for a Kabbe condensation en route to the synthesis of enantioenriched (+)-Cochlearol B
10.
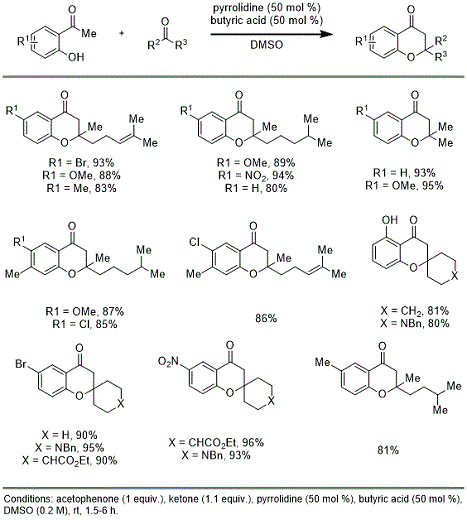
Table 1. Scope of Organocatalyzed Kabbe Condensation (Bapodra 2020)
In this report, we apply this Kabbe condensation protocol from our recent work in an operationally facile and scalable synthesis of chromanone 3. All reagents are commercially available and low cost, making this a readily accessible means of producing large quantities of chromanone compound. This reaction was carried out on a 2 g-scale using catalytic butyric acid and excess pyrrolidine as co-additives. The reported procedure furnishes product in good yield and high purity. This work illustrates the applicability of the Kabbe condensation for scalable synthesis of sought-after chromanone derivatives.
Appendix
Chemical Abstracts Nomenclature (Registry Number)
2'-Hydroxy-5'-methoxyacetophenone; (1) (705-15-7)
6-Methyl-5-hepten-2-one; (2) (110-93-0)
Pyrrolidine; (123-75-1)
Butyric acid; (107-92-6)
DMSO: dimethyl sulfoxide; (67-68-5)

|
Stephen Chamness received his B. Sc. in chemistry from Rice University in Houston, Texas in 2021. He joined Prof. Corinna Schindler's group in 2022 where he is currently working on visible light-mediated energy transfer and photoredox catalysis towards the synthesis of strained ring systems for his doctoral studies. |

|
Emily Traficante received her B.A. in Biochemistry and Molecular Biology from Boston University in 2021. She joined Prof. Corinna Schindler's group in 2022 where she is currently working on the total synthesis of complex natural products for her doctoral studies. |

|
Trenton Vogel received his B. Sc. in chemistry from Saginaw Valley State University in 2020. He joined Prof. Corinna Schindler's group in 2021 where he is currently working on the total synthesis of complex polycyclic natural products for his doctoral studies. |

|
Corinna Schindler started her independent career at the University of Michigan in Ann Arbor, US in 2013. Her research group focuses on the development of new synthetic methods and complex molecule synthesis for biological applications. She was promoted to Associate Professor in 2019 and to Full Professor in 2021. Since 2024, she has been a Canada Research Chair at the University of British Columbia, Vancouver, Canada. |

|
Max Neumann received his B.Sc. in Chemistry and Biochemistry and his M.Sc. in Chemistry from Ludwig-Maximilians-Universität München, where he conducted research with Professors Paul Knochel and Oliver Trapp. He joined Professor Tehshik Yoon's laboratory at the University of Wisconsin-Madison in Fall 2022, where he is currently working on asymmetric intramolecular [2+2] photocycloaddition reactions. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved