Org. Synth. 2025, 102, 367-380
DOI: 10.15227/orgsyn.102.0367
Synthesis of α-Aryl Ketones via the Nickel-Catalyzed Hydroacylation of Aryl Alkenes with Aroyl Fluorides
Submitted by Yoonho Lee,
† Yujin Jung,
† Seonhwa Choo, and Kwangmin Shin*
1Checked by Nicholas Cowper and Nathan Ide
1. Procedure (Note 1)
4-(1-Oxo-1-phenylpropan-2-yl)phenyl acetate (3). A 250-mL, single-necked, round-bottomed flask (24/40 joint) containing a 3 x 1.5-cm Teflon coated egg-shaped magnetic stir bar was oven-dried overnight (130 ℃) and brought into a nitrogen-filled glovebox (Note 2). The flask is charged sequentially with nickel (II) chloride ethylene glycol dimethyl ether complex (494 mg, 2.25 mmol, 0.15 equiv) (Note 3) and potassium phosphate tribasic (9.55 g, 45.0 mmol, 3.0 equiv) (Note 4). The flask is then sealed with a rubber septum, removed from the glovebox, connected to a Schlenk line with a needle adapter, and purged with a nitrogen atmosphere (Figure 1A). Anhydrous N,N-dimethylformamide (30 mL) (Note 5) is added by a 30-mL disposable syringe to the flask. The resulting light-blue colored mixture is stirred (500 rpm) at room temperature (21-22 ℃) for 10 min (Figure 1B). While the solution is stirred, triethoxysilane (3.0 mL, 16.5 mmol, 1.1 equiv) (Note 6) is subsequently added dropwise over 3 min via a 3-mL disposable syringe. The resulting suspension is allowed to stir at room temperature (21-22 ℃), at which time the color of the solution gradually changes to dark blue (Figure 1C).

Figure 1. A. Reaction set up of the flask charged with catalyst and base; B. Reaction mixture after addition of N,N-dimethylformamide; C. After addition of triethoxysilane
After 5 min, 4-acetoxystyrene (2.3 mL, 15.0 mmol, 1.0 equiv) (Note 7) and benzoyl fluoride (2.4 mL, 22.5 mmol, 1.5 equiv) (Note 8) are added in order dropwise over 3 min using 3-mL disposable syringes. An additional 7.5 mL of N,N-dimethylformamide is added via a 12-mL disposable syringe to rinse the flask. The resulting reaction mixture is stirred (500 rpm) at room temperature for 24 h (Figure 2).
Figure 2. Color change over the progress of the reaction: A. Reaction mixture at the beginning; B. After 3 h; C. After 24 h
After being stirred for 24 h, the completion of the reaction is monitored by TLC (Note 9). The septum is then removed, and saturated aqueous sodium bicarbonate (50 mL) is slowly added over 3 min (this results in a strong exotherm, and the mixture should be allowed to cool to room temperature (21-22 ℃) before proceeding) followed by the addition of diethyl ether (50 mL) (Note 10) (Figure 3A). The resulting mixture is allowed to further stir (500 rpm) at room temperature (21-22 ℃) for 20 min (Note 11) (Figure 3B).
Figure 3. A. After addition of saturated aqueous sodium bicarbonate solution and diethyl ether; B. After stirring for 20 min
At this time, the resulting mixture is transferred to a 500 mL separatory funnel. The reaction flask is rinsed with diethyl ether (50 mL), and the washings are added to the separatory funnel (some tar residue remained in the flask and could not be transferred). The funnel is shaken, and the layers are separated (Figure 4A). The aqueous layer was extracted with diethyl ether (2 x 50 mL). The organic layers are then combined, dried over sodium sulfate (40 g) (Note 12), and vacuum-filtered using a 110 mL, 10 μm polypropylene fritted filter funnel into a 500 mL round-bottomed flask (Figure 4B). The resulting solution is concentrated under reduced pressure with the aid of a rotary evaporator (35 ℃ water bath temperature, 30 mm Hg) to afford the concentrated crude mixture as a yellow oil (Figure 4C).
Figure 4. A. Extraction set up; B. Filtration set up; C. Concentrated crude reaction mixture
A flash column (5.5 cm diameter x 23 cm height) is wet packed with silica gel (150 g) (Note 13) in hexane (Note 14). The crude mixture is loaded onto the stationary phase. The flask containing the crude mixture is rinsed with ethyl acetate in hexane (1/5, v/v, 2 x 5 mL) (Note 15), and the washings are also loaded onto the column. Once adsorbed, sea sand (Note 16) is added to protect the silica layer (Figure 5A). The column is then eluted with a gradient of ethyl acetate in hexane (Note 17). The product-containing fractions 34-75 are combined in a 1-L round-bottomed flask and concentrated under reduced pressure with the aid of a rotary evaporator (35 ℃ water bath temperature, 30 mm Hg) to afford a white solid. This solid is further dried under high vacuum (8 mm Hg, room temperature (21-22 ℃)) for 5 h to afford (Run 1: 2.64 g, 62%) (Run 2: 2.65 g, 62% yield) 4-(1-Oxo-1-phenylpropan-2-yl)phenyl acetate (3) as a white powder (Notes 18-19) (Figure 5B).
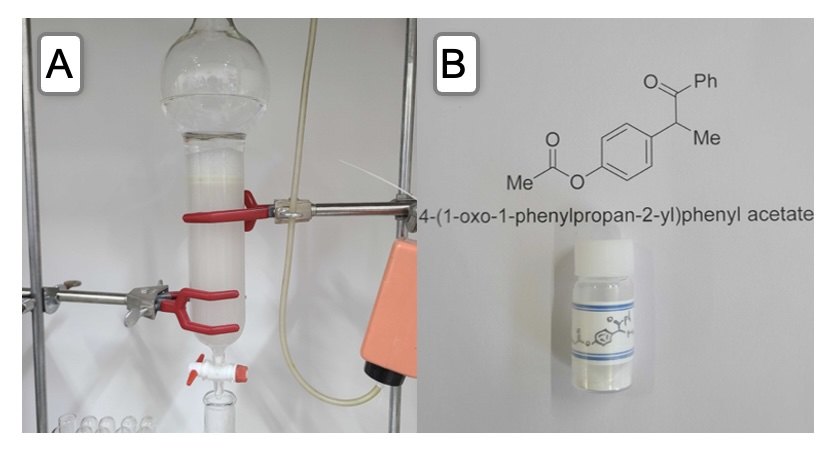
Figure 5. A. Purification set up; B. Isolated title compound
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of “Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also “Identifying and Evaluating Hazards in Research Laboratories” (American Chemical Society, 2015) which is available via the associated website “Hazard Assessment in Research Laboratories” at
https://www.acs.org/about/governance/committees/chemical-safety.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
nickel(II) chloride ethylene glycol dimethyl ether complex,
potassium phosphate tribasic,
N,N-dimethylformamide,
triethoxysilane,
4-acetoxystyrene,
benzoyl fluoride,
diethyl ether,
ethyl acetate,
hexane,
1,1,2,2-tetrachloroethane,
sodium sulfate, and silica gel.
Triethoxysilane, listed by various vendors as a H315 (Category 2 Causes skin irritation) or as a H318 (Category I Causes Serious Eye Damage). After the reaction, work-up process should be carried out prior to any subsequent manipulations to ensure complete quenching of the residual
triethoxysilane.
2. A glove box is used for the storage and handling for the hygroscopic reagents such as
nickel (II) chloride ethylene glycol dimethyl ether complex,
potassium phosphate tribasic and
triethoxysilane.
3.
Nickel (II) chloride ethylene glycol dimethyl ether complex (97%) was purchased from Strem, stored in the
nitrogen-filled glovebox, and used as received.
4.
Potassium phosphate tribasic (reagent grade, ≥98%) was purchased from Sigma-Aldrich, stored in the
nitrogen-filled glovebox, and used as received.
5.
N,N-Dimethylformamide (anhydrous, 99.8%) was purchased from Sigma-Aldrich in Sure-Seal
TM bottle and used as received.
6.
Triethoxysilane (95%) was purchased from Sigma-Aldrich, stored in the
nitrogen-filled glovebox, and used as received. A notable decrease in yield was observed when
triethoxysilane stored outside of a glovebox was used.
7.
4-Acetoxystyrene (96%) was purchased from Thermo Scientific and used as received. The checkers used
4-acetoxystyrene (96%) purchased from Sigma-Aldrich.
8.
Benzoyl fluoride (>98%) was purchased from TCI and used as received.
9. The reaction progress was monitored by TLC analysis,
ethyl acetate in
hexane (1/5, v/v), R
f value of starting material (compound 1) = 0.59, R
f value of the title compound (
3) = 0.25
Figure 6. TLC analysis of the reaction mixture: A. Visualized by 254 nm UV. Left to right: Starting material (compound 1); Co-spot of starting material (compound 1) and the reaction mixture; The reaction mixture B. Visualized by phosphomolybdic acid (PMA) stain. Left to right: Starting material (compound 1); Co-spot of starting material (compound 1) and the reaction mixture; reaction mixture
11. After quenching the mixture was stirred for additional 20 min to ensure the destruction of any remaining
triethoxysilane and to promote phase separation.
12.
Sodium sulfate (Anhydrous, 99%) was purchased from Samchun Pure Chemical (Korea) and used as received.
13. Silica gel (40-63 μm, 230-400 mesh, 60 Å pore diameter) was purchased from ZEOCHEM and used as received.
14.
Hexane (85%) was purchased from Daejung Chemical & Metals (Korea) and used as received.
15.
Ethyl acetate (99%) was purchased from Daejung Chemical & Metals (Korea) and used as received.
16. Sea sand (10-20 mesh) was purchased from Yakuri Pure Chemicals (Japan) and used as received.
17. Column chromatography was performed as follows: A flash column (5.5 cm column diameter x 23 cm height) is wet packed with silica gel (150 g) and
hexane (400 mL) to give a column height of 19 cm. The crude mixture is loaded onto the column using 10 mL of
ethyl acetate in
hexane (1/5, v/v). Then, sea sand (3 cm height) is added to the top of the column. The fractions are collected in 16 x 150 mm test tubes (ca. 10 mL per fraction). Elution is begun with 200 mL of
hexane, then continued with 205 mL of
ethyl acetate in
hexane (1/40, v/v), then 210 mL of
ethyl acetate in
hexane (1/20, v/v), then 220 mL of
ethyl acetate in
hexane (1/10, v/v), then 1.2 L of
ethyl acetate in
hexane (1/5, v/v) using compressed air. The checkers found that column chromatography performance improved with decreasing column pressures.
Figure 7. TLC analysis of column chromatography visualized by 254 nm UV
18. Characterization data of
4-(1-oxo-1-phenylpropan-2-yl)phenyl acetate (
3):
1H NMR
pdf (400 MHz, CDCl
3) δ 7.98-7.89 (m, 2H), 7.54-7.44 (m, 1H), 7.42-7.36 (m, 2H), 7.32-7.27 (m, 2H), 7.06-6.96 (m, 2H), 4.70 (q,
J = 6.9 Hz, 1H), 2.26 (s, 3H), 1.52 (d, J = 6.9 Hz, 3H).
13C NMR
pdf (101 MHz, CDCl
3) δ: 200.2, 169.4, 149.5, 138.8, 136.3, 132.9, 128.8, 128.7, 128.6, 122.0, 47.1, 21.1, 19.5. IR (Film): 2985, 2938, 1759, 1677, 1596, 1584, 1500, 1458, 1449, 1420, 1368, 1341, 1323, 1303, 1255, 1215, 1191, 1167, 1109, 1078, 1046, 1013, 955, 924, 908, 855, 819, 771, 748, 718, 699, 682, 659, 623 cm
-1. HRMS (ESI+): exact mass calc′d. for C
17H
17O
3+ [M+H]
+: 269.11722; found: 269.11659 . Melting point: 45-48 ℃. The product (
3) is stable on the benchtop at room temperature (21-22 ℃) under air atmosphere.
19. The purity of the title compound (
3) was determined to be 95% for Run 1 and 98% for Run 2 by quantitative
1H NMR
pdf spectroscopy in CDCl
3 using 20 mg of the product (
3) and 19.33 mg of
1,1,2,2-tetrachloroethane (Sigma-Aldrich, >98%, used as received) as an internal standard.
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Owing to the prevalence of α-aryl ketones in natural products, biologically active compounds and synthetic building blocks, considerable effort has been devoted to developing efficient synthetic methods for their preparation.
2 Among the synthetic strategies developed, the reaction between aryl alkenes with acyl electrophiles under transition metal-hydride catalysis, commonly referred to as hydroacylation, stands out as one of the most straightforward synthetic routes. Based on the pioneering report by Miura,
3 in 2006, Krische developed a rhodium hydride-catalyzed branch-selective hydroacylation of activated olefins including aryl alkenes with acid anhydrides.
4 In 2016, the Buchwald group reported an enantioselective variant of this transformation by using copper-hydride catalysis.
5 They later expanded this chemistry to hydroacylation of aryl alkenes with unsaturated carboxylic acids as acyl group donors.
6 Zhu and co-workers developed a nickel hydride-catalyzed migratory hydroacylation of olefins, using acid anhydrides generated in-situ from carboxylic acids.
7 Despite these advances, however, there is still an ongoing need for the development of an alternative hydroacylation method capable of employing other types of acyl electrophiles.
Acyl fluorides have been employed as efficient acyl group donors in various organic transformations.
8 Their use in transition metal catalysis was first demonstrated by the Rovis group in 2004.
9 In this pioneering work, they employed acyl fluorides as acyl sources for a nickel-catalyzed Negishi-type cross-coupling reaction. In 2019, Iwai and Sawamura described a hydroacylation of aryl alkenes with aroyl fluorides through a dual copper and nickel catalysis.
10 While notable, this hydroacylation protocol necessitates a bespoke phosphine ligand system, and the scope of olefin is limited to terminal aryl alkenes. In this context, in 2022, our group reported a nickel-catalyzed hydroacylation of terminal and internal aryl alkenes with aroyl fluorides.
11 Key features of this approach include the use of single nickel catalyst and the absence of the need for an exogeneous ligand.
This hydroacylation method exhibits broad substrate scope (Table 1). Aryl alkenes substituted with various functional groups such as methoxy, chloro, pinacolborane, ester, and amide could be well-tolerated. In addition, the protocol is compatible with heterocyclic groups, including dibenzofuran, indole and pyridine. Along with terminal styrenes, internal aryl alkenes could also participate in this transformation. Acyl fluorides bearing various electronically different substituents could be efficiently converted into the desired products. This approach can be applied to late-stage derivatization of complex molecules. Finally, the method is amenable to a gram-scale hydroacylation, as described in this report.
Table 1. Selected reaction scope of the NiH-catalyzed hydroacylation
Yields of isolated products are shown. (a) The reaction was performed at 0.7 M in DMF solvent. (b) K3PO4 (50 mol%) was used. (c) 2.0 equiv of aryl alkene and 1.0 equiv of aroyl fluoride were used. (d) 3.0 equiv of (EtO)3SiH was used. (e) 2.0 equiv of K3PO4 was used. (f) The reaction was performed at 0.4 M in DMF solvent. (g) 2.5 equiv of aroyl fluoride was used.
Appendix
Chemical Abstracts Nomenclature (Registry Number)
Nickel (II) chloride ethylene glycol dimethyl ether complex; (29046-78-4)
Potassium phosphate tribasic; (7778-53-2)
N,N-Dimethylformamide; (68-12-2)
Triethoxysilane; (998-30-1)
4-Acetoxystyrene; (2628-16-2)
Benzoyl fluoride; (455-32-3)
Diethyl ether; (60-29-7)
Ethyl acetate; (141-78-6)
Hexane; (110-54-3)
1,1,2,2-Tetrachloroethane; (79-34-5)
Sodium sulfate; (7757-82-6)

|
Yoonho Lee was born in 1999 in South Korea. He earned his B.S. degree in chemistry from Hanyang University ERICA in 2022. He is currently pursuing an integrated master's and Ph.D. degree in the department of chemistry at Sungkyunkwan University (SKKU) under the supervision of Prof. Kwangmin Shin. His research interests focus on the development of nickel-hydride catalyzed hydrofunctionalization of olefins with various electrophiles. |

|
Yujin Jung was born in 2000 in South Korea. She earned her B.S. degree in chemistry from Kyonggi University in 2023. She is currently pursuing a master's degree in the department of chemistry at Sungkyunkwan University (SKKU) under the supervision of Prof. Kwangmin Shin. Her research focuses on the development of first-row transition metal-hydride catalysis for hydroacylation of various unsaturated systems. |

|
Seonhwa Choo was born in 1999 in South Korea. She earned her B.S. degree in chemistry from Chungbuk National University in 2024. She is currently pursuing a master's degree in the department of chemistry at Sungkyunkwan University (SKKU) under the supervision of Prof. Kwangmin Shin. Her research focuses on the development of transition metal hydride- catalyzed enantioselective olefin hydro-functionalization. |

|
Kwangmin Shin received his B.S. and Ph.D. degrees in chemistry from Korea Advanced Institute of Science and Technology (KAIST) under the supervision of Prof. Sukbok Chang in 2012 and 2017, respectively. After posdoctoral research with Prof. Stephen L. Buchwald at MIT, he began his independent career at Sungkyunkwan University (SKKU) as an assistant professor in 2020. His current research interests encompass transition metal-hydride catalysis, enantioselective catalysis, carbon-fluorine bond formation/cleavage for the synthesis of value-added compounds. |

|
Nicholas Cowper is a Senior Scientist in the Process Chemistry group at Abbvie. He received his BSc from Queen’s University and his PhD from CalTech working with Sarah Reisman on strategies to access 1,2-oxazine natural products. Following his graduate studies he moved to UW-Madison to work with Zach Wickens where he developed electron-primed photocatalytic transformations. Nicholas joined Abbvie in 2021. Along with his pipeline work at AbbVie, Nicholas works in the Center of Catalysis developing platform tools to implement non-precious metal catalysis, photochemistry, and electrochemistry. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved