Org. Synth. 2025, 102, 398-413
DOI: 10.15227/orgsyn.102.0398
Large-Scale Alkene Dimesyloxylation
Submitted by Shyam Sathyamoorthi*
Checked by Hin Yu Kenneth Huang, Yu Chen, and Pauline Chiu
1. Procedure (Note 1)
Octane-1,2-diyl dimethanesulfonate (1). A 250-mL Erlenmeyer flask was equipped with a Teflon-coated magnetic stir bar (4 x 0.75 cm, rod-shaped). The flask was charged with 1-octene (1.12 g, 10.0 mmol, 1.0 equiv) (Note 2) using a pipette. Dichloromethane (150 mL) (Note 3) was measured in a graduated cylinder and added to the flask. The flask was placed in an ice-water bath on a magnetic stir plate, and stirring (400 rpm) was commenced. After cooling for 10 min to 0 ℃ (checked using a thermometer), Iodomesitylene diacetate (4.01 g, 11.0 mmol, 1.1 equiv) (Note 4) was added to the reaction mixture. The reaction mixture was stirred in an ice-water bath for an additional 5 min. Following this time, methanesulfonic acid (1.4 mL, 2.1 g (calculated), 21.8 mmol, 2.2 equiv) (Note 5) was added using a plastic syringe fitted with a 20G needle. The flask was closed using a polyethylene cap. The color of the reaction mixture turned from colorless to yellow immediately upon the addition of methanesulfonic acid (Figure 1A and 1B). The ice-water bath was removed, and the reaction mixture was allowed to stir at room temperature (~22 ℃) for 2 h (Note 6), over which time the color faded from markedly yellow to a very pale yellow (Figure 1B and 1C).

Figure 1. A. Reaction mixture prior to addition of methanesulfonic acid; B. Reaction mixture after adding methanesulfonic acid; C. After ~ 2 h at room temperature, the yellow color fades to a barely noticeable, pale-yellow hue (Photos provided by the checkers)
The reaction mixture was diluted with CH2Cl2 (50 mL) (Note 3) and transferred to a 1-L separatory funnel. Saturated, aqueous Na2S2O3 solution (Note 7) was added, followed by 75 mL of water. The reaction mixture was shaken vigorously and allowed to separate (Note 8). The bottom, organic layer was drained and the upper aqueous layer was back-extracted with 50 mL of CH2Cl2. The collected organic layers were combined, returned to the separatory funnel, and then dried by shaking with brine (50 mL) (Note 9). The bottom, organic layer was drained, dried with 25 g of anhydrous MgSO4 (Note 10), and filtered through a fritted Buchner funnel (150 mL, medium porosity) under house vacuum (~75 mm Hg) into a 500-mL round-bottom flask (24/40 joint). The MgSO4 filter cake was rinsed with an additional 100 mL of CH2Cl2. The filtrate was concentrated on a rotary evaporator (Note 11). The resulting yellow oil (Figure 2A) was analyzed by TLC (Notes 12 and 13, Figure 2B, 2C) and 1H NMR spectroscopy and found to be a mixture comprised mainly of product 1 and mesityl iodide.
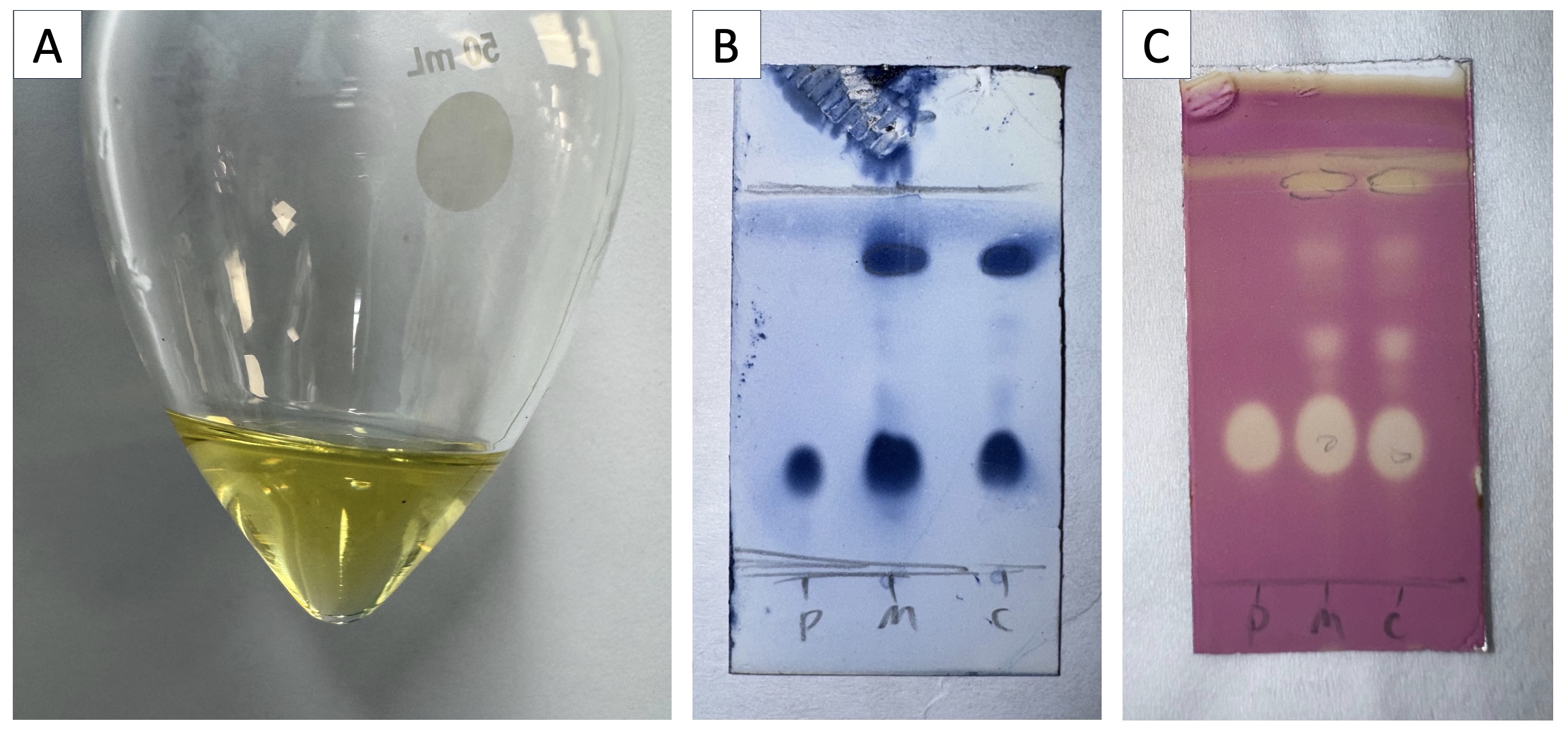
Figure 2. A. Crude product 1 was obtained as a yellow oil; B. TLC analysis of the reaction mixture after workup, stained with CAM stain (Note 13); C. TLC analysis of the reaction mixture after workup, stained with KMnO4 stain (Note 14). Mobile phase: 1:2 EtOAc/hexanes. P = authentic product 1, from a prior reaction (Rf = 0.3), M = co-spot of 1 and crude reaction mixture, C = crude reaction mixture. (Photos provided by the checkers)
This residue was purified by flash column chromatography on silica gel (Figure 3A) (Notes 15,16,17,18). The fractions containing pure product 1 were pooled and concentrated under reduced pressure on a rotary evaporator (Note 11). The resulting oil (Figure 3B) was rid of residual volatiles by applying a high vacuum (~0.5 mm Hg) overnight. Compound 1 was obtained as a pale-yellow oil (2.24 g, 7.41 mmol, 74% yield) (Note 19, 20).
Figure 3. A. Flash silica gel column chromatography set-up; B. Compound 1 (octane-1,2-diyl dimethanesulfonate) is a pale-yellow oil. (Photo provided by the checkers)
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of “Prudent Practices in the Laboratory” (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also “Identifying and Evaluating Hazards in Research Laboratories” (American Chemical Society, 2015) which is available via the associated website “Hazard Assessment in Research Laboratories” at
https://www.acs.org/about/governance/committees/chemical-safety.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with the chemicals used in this manuscript, as well as the proper procedures for product preparation.
2.
1-Octene (99%) was purchased from Thermo Scientific Chemicals and used directly by both the authors and the checkers.
3.
Methylene chloride (HPLC Grade, 99.7%, stabilized with amylene) was purchased from Fisher Scientific and used directly. The checkers obtained
methylene chloride (AR Grade, 99.8%,) from RCI Labscan Ltd., which was used as received.
4.
Iodomesitylene diacetate (97%) was purchased from ChemScene and used directly. The checkers obtained
iodomesitylene diacetate (98%) from Shanghai Macklin Biochemicals Co. Ltd., which was used as received.
5.
Methanesulfonic acid (99%) was purchased from Sigma-Aldrich and used directly. The checkers obtained
methanesulfonic acid (99%, extra pure) from Thermo Scientific Chemicals, which was used as received.
6. Monitoring of the reaction progress using thin layer chromatography was not feasible, as
1-octene at the concentration used in the reaction could hardly be visualized by UV or by staining. The checkers followed the reaction progress by
1H NMR spectroscopy, which indicated that most of the
1-octene was consumed after 2 h at room temperature (22 ℃).
7.
Sodium thiosulfate pentahydrate (>99%) was purchased from Fisher Scientific and used as received. The checkers obtained
sodium thiosulfate (>98%) from Thermo Scientific Chemicals, which was used as received.
8. When the reaction mixture is shaken with saturated aqueous
Na2S2O3 solution, the
CH2Cl2 layer is the top layer (Figure 4A). An emulsion forms from shaking this mixture which makes a clean separation of layers difficult. The addition of 75 mL of water to dilute the
Na2S2O3 solution changes the density of the aqueous layer, causing the organic
CH2Cl2 layer to be at the bottom, and the demarcation of the two layers is clearer (Figure 4B).
Figure 4. A. Saturated aqueous Na2S2O3 solution and CH2Cl2 mixture in a separatory funnel after being shaken and prior to dilution with water. B. Mixture in the separatory funnel after additional dilution with 75 mL water. (Photos provided by the checkers)
9. Brine (saturated aqueous solution of
NaCl) was made by saturating deionized
H2O with
sodium chloride (99%) which was purchased from Fisher Scientific and used directly. The checkers obtained
sodium chloride (AR Grade) from Dieckmann (Hong Kong) Chemical Industry Co. Ltd., which was used as received.
10.
Magnesium sulfate (anhydrous) was purchased from Fisher Scientific and used directly. The checkers obtained anhydrous
magnesium sulfate from Dieckmann Chemical Industry Co. Ltd. (Hong Kong), which was used as received.
11. The rotovap was operated at ~100 mm Hg and a water bath temperature of ~30 ℃.
12. Thin layer chromatography (TLC) was performed on silica gel 60 F254 (glass-backed TLC plates). The checkers used silica gel 60 F254 aluminum-backed TLC plates, purchased from Millipore. Crude product
1 obtained as a yellow oil.
1H NMR
pdf analysis shows that it is a mixture of product
1 and mesityl iodide. The authors remarked that this unpurified residue darkens to a brown color upon standing, but this color change was not observed by the checkers.
13.
Cerium ammonium molybdate (CAM) stain was prepared by combining 5 g ammonium molybdate, 1 g cerium sulfate, and 10 mL concentrated sulfuric acid in 90 mL of deionized
H2O. TLC plates are visualized after dipping in the stain and heating with a heat gun.
14.
KMnO4 stain was prepared by dissolving 1.5 g
KMnO4, 10 g
K2CO3, and 1.25 mL of 10% aqueous
NaOH solution in 200 mL of deionized
H2O. TLC plates are visualized after dipping in the stain and gently heating with a heat gun.
15. For flash column chromatography, a Synthware column (Item number: C184463, with column length of 30.5 cm and internal diameter of 4 cm) was used. This column was dry-packed with 105 g of silica gel and equilibrated using 300 mL of hexanes (column was ~7 inches in height after equilibration). The residue was loaded on the column neat and allowed to adsorb onto the silica gel by applying compressed air. An additional 2 mL of
CH2Cl2 was used to rinse the flask and this was transferred to the column. The walls of the column were rinsed with 1 mL of
CH2Cl2. After full adsorption of these rinses onto the silica gel, the top of the silica gel column was layered with sand (purchased from Shanghai Macklin Biochemicals Co. Ltd., 40-60 mesh, AR grade) to a thickness of ~1.5 cm. The column was flushed with 100 mL of hexanes, then 2.0 L of 15%
EtOAc/hexanes. The column was then eluted with 3.25 L of 20%
EtOAc/hexanes, while the eluent was collected as 40 mL fractions. The fractions were checked by TLC (1:2
EtOAc:hexanes) and visualized using CAM stain (Figure 5). In total, 63 test-tube fractions of ~40 mL each were collected.
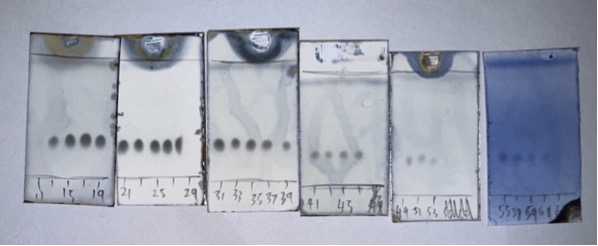
Figure 5. Fractions 11-55 containing pure compound 1 were collected. (Photos provided by the checkers)
16.
Ethyl acetate (Certified ACS Reagent, 99.5%) was purchased from Fisher Scientific and used directly. The checkers purchased
ethyl acetate (GR grade) from Duksan Pure Chemicals Ltd., which was purified by distillation on a rotary evaporator before use.
17. Hexanes (98.5%) was purchased from Fisher Scientific and used directly. The checkers purchased hexanes (95%, GR grade) from Duksan Pure Chemicals Ltd., which was distilled on a rotary evaporator before use.
18. Silica gel (grade 60, 230-400 mesh) was purchased from Fisher Scientific. The checkers purchased silica gel (grade 60, 0.040-0.063 mm, 230-400 mesh ASTM) from Millipore.
19. Analytical data for
octane-1,2-diyl dimethanesulfonate (
1):
1H NMR
pdf (600 MHz, CDCl
3) δ 4.86 (dtd,
J = 8.0, 5.9, 2.8 Hz, 1H), 4.38 (dd,
J = 11.5, 2.9 Hz, 1H), 4.26 (dd,
J = 11.5, 6.4 Hz, 1H), 3.08 (s, 3H), 3.07 (s, 3H), 1.77 (dddd,
J = 14.2, 10.2, 7.7, 5.4 Hz, 1H), 1.73 - 1.65 (m, 1H), 1.51 - 1.16 (m, 8H), 0.87 (t,
J = 7.0 Hz, 3H) ppm;
13C NMR
pdf (101 MHz, CDCl
3) δ 79.4, 69.7, 38.9, 37.9, 31.6, 31.3, 28.9, 24.9, 22.6, 14.1 ppm; IR (neat) ν 2931, 2859, 1460, 1332, 1169, 970, 907, 812, 524 cm
-1; HRMS (ESI) m/z = [M + Na
+] Calc'd C
10H
22NaO
6S
2+ 325.0750; Found 325.0750. The purity of
1 was determined to be 98% by qNMR
pdf using
p-nitrotoluene (99.5%, purchased from Aladdin Scientific) as an internal standard.
20. A second full scale reaction performed by the checkers provided 2.22 g (74% yield) of compound
1 of 98% purity as determined by qNMR.
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Compared to alkene dihydroxylation,
2,3 alkene disulfonoxylation is an under-explored technology. Our efforts were inspired by pioneering contributions from Koser and Zhandkin.
4,5,6 Despite their important advances, we identified several drawbacks in literature protocols of alkene disulfonoxylation, including the use of non-commercial iodine (III) sources, the necessity of excess alkene substrate, and a narrow substrate scope with respect to both the alkene substrate and the sulfonic acid partner. At the outset of our investigations, it was our goal to develop a disulfonoxylation reaction that used inexpensive, commercial reagents and in which the alkene substrate was the limiting reagent. It was a priority that our protocol be compatible with a range of sulfonic acids, as most literature examples were ditosyloxylations.
Schemes 1 and
2 describe the scope of our reaction,
7 a scaled-up version of which is the topic of this Organic Syntheses contribution. A variety of sulfonic acids engaged productively in this oxidation reaction. Several aryl halides were tolerated, and electron rich moieties such as methoxy groups survived (
Scheme 1, Entry 1). With many of these sulfonic acids, good product yields could be achieved using either PhI(OAc)
2 or iodomesitylene diacetate. With “bulkier” sulfonic acids (
Scheme 1, Entries 2, 4, 5, and 6), better results were achieved with the more electrophilic PIFA. Alkyl sulfonic acids were also very compatible (
Scheme 1, Entries 3 and 5).
Several alkene substrates were tolerated by our optimized conditions (Scheme 2). The reactions were quite chemoselective, and even substrates with benzylic C-H bonds gave the expected disulfonoxylated products in good yields (Scheme 2, Entries 1 and 4). Where relevant, the reactions were stereospecific (within the limits of 1H NMR detection), and we have assigned the relative stereochemistry by analogy to compound 15 (CCDC 2332152). Nitrile and acetate functionalities were well tolerated (Scheme 2, Entry 5). Overall, our protocols work best with terminal alkenes, but certain disubstituted cyclic alkenes give products in reasonable yields. Some poorly performing substrates include cyclopentene, 1-methyl-1-cyclohexene, cis-4-octene, and trans-4-octene.
Scheme 1. Survey of Sulfonic Acid Coupling Partners
Scheme 2. Alkene scope and functional group compatibility
Under our reaction conditions, some substrates gave unexpected products (
Scheme 3). The most controversial of these products were those arising from styrenyl substrates. Some literature reports have claimed that these products are the expected vicinal disulfonates.
4,8,9,10 Others have disputed these results and have suggested that the products are geminally disulfonoxylated!
11 We have obtained crystal structures of these compounds and have unambiguously shown that these are in fact geminally disulfonoxylated.
Scheme 3. Substrates which give interesting and unexpected products
We hypothesize that an unusual phenonium rearrangement
12 underlies product selectivity with these styrenyl substrates (
Scheme 4). Literature reports have focused on chalcone substrates, but we have demonstrated that this rearrangement is competent for cinnamyl esters as well (
Scheme 3).
Scheme 4. Proposed mechanisms and some relevant experiments
Appendix
Chemical Abstracts Nomenclature (Registry Number)
Oct-1-ene: (111-66-0)
Iodomesitylene diacetate: (33035-41-5)
Methanesulfonic acid: (75-75-2)

|
Shyam Sathyamoorthi completed a B.S. degree in Cell and Molecular Biology with a minor in Chemistry at Tulane University, New Orleans, Louisiana, where he worked in the labs of Professor Ken Muneoka and Professor Robert A. Pascal, Jr. He then completed a PhD in chemistry at Stanford University under the guidance of Professor Richard N. Zare (2018) as well as a Doctor of Medicine degree at the Stanford University School of Medicine (2019). In July 2019, he started his independent career as an assistant professor in the Department of Medicinal Chemistry at the University of Kansas, Lawrence, KS, USA. He was promoted to associate professor (with tenure) in 2024. |

|
Hin Yu Kenneth Huang is from Hong Kong, P. R. China. He is a senior undergraduate at Occidental College and worked with Professor Jeffery Cannon on photocatalytic reductions of aromatic ketones. He is currently taking a gap semester and working as a student research assistant in the laboratory of Professor Pauline Chiu at the University of Hong Kong. |

|
Yu Chen was born in Anhui, Mainland China. She received her B.Sc. in 2013 from Anhui University. In 2020, she completed her Ph.D. at the University of Hong Kong under the supervision of Professor Pauline Chiu. Currently, she is pursuing postdoctoral research at the University of Hong Kong, focusing on organic synthesis and medicinal chemistry. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved