Org. Synth. 2025, 102, 414-427
DOI: 10.15227/orgsyn.102.0414
Acid-Mediated Hydroaminomethylation of Unactivated Alkenes
Submitted by Uroš Vezonik,
1 Paolo Piacentini
1 and Nuno Maulide*
1Checked by Anagha Veluthanath Nair and M. Kevin Brown
1. Procedure (Note 1)
A. N,N,N',N'-Tetrabenzylmethanediamine (3). A 250-mL round-bottom flask (NS 29) is equipped with a 3-cm magnetic stir bar and charged with dibenzylamine (35.3 mL, 180 mmol, 2.00 equiv) (Note 2). To this, 45 mL of distilled water is added, and the flask is sealed with a septum. The flask containing the mixture is placed into an ice/water bath at 0 ℃ and, after stirring for 5 min, an aqueous solution of formaldehyde (37%, 6.70 mL, 90.0 mmol, 1.0 equiv) (Note 3) is added dropwise over 5 min, using a syringe (with no pressure-equalizing measures necessary) (Figure 1A). The resulting biphasic mixture is stirred vigorously (1000 rpm) at room temperature (23 ℃) for 10 min, during which time a suspension forms (Figure 1B). The septum is subsequently removed, and, to the suspension, 100 mL of ethyl acetate (EtOAc) (Note 4) is added. The flask is placed in an ultrasonic bath and sonicated for 10 min at 23 ℃ to assist with mixing and dissolution. After sonication, the heterogeneous mixture is further stirred (750 rpm) for an additional 10 min at 23 ℃ until the solid has completely dissolved (Figure 1C).

Figure 1. A. Addition of formaldehyde solution at 0 ℃; B. Suspension after stirring for 10 min at 23 ℃; C. Organic and aqueous phases after sonication and further stirring.
The aqueous and organic phases are transferred to a 500-mL separatory funnel. The round-bottom flask is washed with ethyl acetate (2 × 20 mL), and the washings are also transferred to the separatory funnel. The organic layer is separated from the aqueous layer, and the aqueous layer is extracted with EtOAc (2 × 50 mL). The organic phases are combined in a 500-mL Erlenmeyer flask, dried over 20 g of anhydrous sodium sulfate (Note 5) and filtered through a cotton plug in a glass funnel into a 1-L round-bottom flask. The filtrate is concentrated under reduced pressure by rotary evaporation (320 mbar at 40 ℃ for 5 min, followed by 180 mbar at 40 ℃ for 25 min). The remaining solution is transferred to a 250-mL round-bottom flask with EtOAc (3 × 20 mL) and again concentrated by rotary evaporation (320 mbar at 40 ℃ for 5 min, followed by 35 mbar at 40 ℃ for 25 min). The resulting solid is further dried under high vacuum (3 × 10-2 mbar) at ambient temperature for 4 h to afford product 3, which was used in the subsequent step without further purification (34.9 g, 95% yield) (Figure 2) (Notes 6, 7).

Figure 2. Dried N,N,N',N'-tetrabenzylmethanediamine.
B. N-Benzyl-5-phenylpentan-1-amine (5). A 500-mL round-bottom flask (NS 29) is charged with N,N,N',N'-tetrabenzylmethanediamine (32.5 g, 80.0 mmol, 4.0 equiv) and a 4-cm long magnetic stir-bar. The flask is then fitted with a pressure-equalizing dropping funnel containing trifluoroacetic acid (33.3 mL, 0.6 M relative to the alkene) (Note 8) and equipped with a septum and an argon balloon. The flask is then partially submerged in an ice/water bath at 0 ℃ and the temperature of the contents is allowed to equilibrate while stirring for 5 min. Then, while vigorously stirring (750 rpm), the trifluoroacetic acid contained in the dropping funnel is added dropwise over 10 min (the rate of addition, 8 drops per second) (Figure 3A). Following addition of trifluoroacetic acid (Figure 3B), the dropping funnel is removed, briefly exposing the reaction to air, and 4-phenyl-1-butene (3.07 mL, 20.0 mmol, 1.0 equiv) (Note 9) is added in two portions using a syringe. A Vigreux column is fitted to the flask, with the top closed using a septum and a nitrogen line. The flask is then placed in a pre-heated oil bath at 75 ℃ (Figure 3C). The reaction mixture initially forms a suspension, which gradually transforms into a clear orange solution upon heating (Figure 3D).

Figure 3. A. Aminal before addition of TFA at 0 ℃; B. After addition of TFA at 0 ℃; C. Reaction set-up after addition of 4-phenyl-1-butene; D. Reaction mixture after 10 min of heating at 75 ℃.
The reaction mixture is vigorously stirred (750 rpm) at 75 ℃ for 15 h, after which it is allowed to cool to room temperature. The volatile components are then removed under reduced pressure using a rotary evaporator (300 mbar at 40 ℃ for 5 min, followed by 35 mbar at 40 ℃ for 10 min) (Figure 4A). The resulting crude mixture is treated with 40 mL of a 1 M aqueous solution of sodium hydroxide (Notes 10, 11) and 20 mL of dichloromethane (Figure 4B) (Note 12). The suspension is then stirred vigorously (750 rpm) at 23 ℃ for 1 h (Figure 4C). After this time, while stirring continues, 5 M aqueous solution of sodium hydroxide (Note 13) is added portion-wise (5 x 20 mL) over 5 min to the reaction mixture (Figure 4D), followed by the addition of 50 mL of dichloromethane (Figure 4E).

Figure 4. A. Reaction mixture after concentration using a rotatory evaporator; B. Reaction mixture after first addition of NaOH (1 M) and dichloromethane; C. Reaction mixture after additional stirring for 1 h; D. Reaction mixture after addition of NaOH (5 M); E. Reaction mixture after addition of a second portion of dichloromethane.
The resulting biphasic mixture is transferred into a 500-mL separatory funnel, and the reaction flask is washed with dichloromethane (3 × 20 mL) (Figure 5A). The organic phase is separated, and the retained aqueous phase is extracted with dichloromethane (3 × 100 mL). The combined organic phases are transferred into an Erlenmeyer flask, dried over 40 g of anhydrous Sodium sulfate (Na2SO4), and filtered through cotton into a 1-L round-bottom flask. The Erlenmeyer flask is rinsed with dichloromethane (3 × 20 mL), and the combined filtrate is concentrated under reduced pressure using a rotary evaporator (650 mbar at 40 ℃ for 30 min, followed by 35 mbar at 40 ℃ for 30 min) to afford the crude product (6.64 g), (Figure 5B). The crude product is then purified by flash column chromatography on silica gel.
Figure 5. A. Separation of organic and aqueous phases; B. Crude product after reduced-pressure concentration.
Column chromatography (Note 14): To the crude product, 10 mL of dichloromethane is added, and the flask is sonicated in an ultrasonic bath for 5 min to ensure thorough dissolution. The sample is then loaded onto a silica gel column (Notes 15, 16), with the flask rinsed with dichloromethane (2 × 5 mL) to ensure complete transfer. Fractions of 18 mL are collected during the chromatography process. The column is first eluted with 10% EtOAc in heptane (1.2 L) (Note 17), followed by 20% EtOAc in heptane (1.2 L), 30% EtOAc in heptane (600 mL), 50% EtOAc in heptane (600 mL) and lastly 100% EtOAc (3 L). Fractions 65 to 150 contain pure product and are combined (Figure 6A) (Note 18), after which the solvent is removed by rotary evaporation (150 mbar at 40 ℃ for 15 min, followed by 10 mbar at 40 ℃ for 45 min). The resulting oil is further dried under a high vacuum (3 x 10-2 mbar at 23 ℃) at ambient temperature for 6 h to obtain product 5 as a pale-yellow oil (4.15 g, 82%) (Figure 6B) (Notes 19, 20).
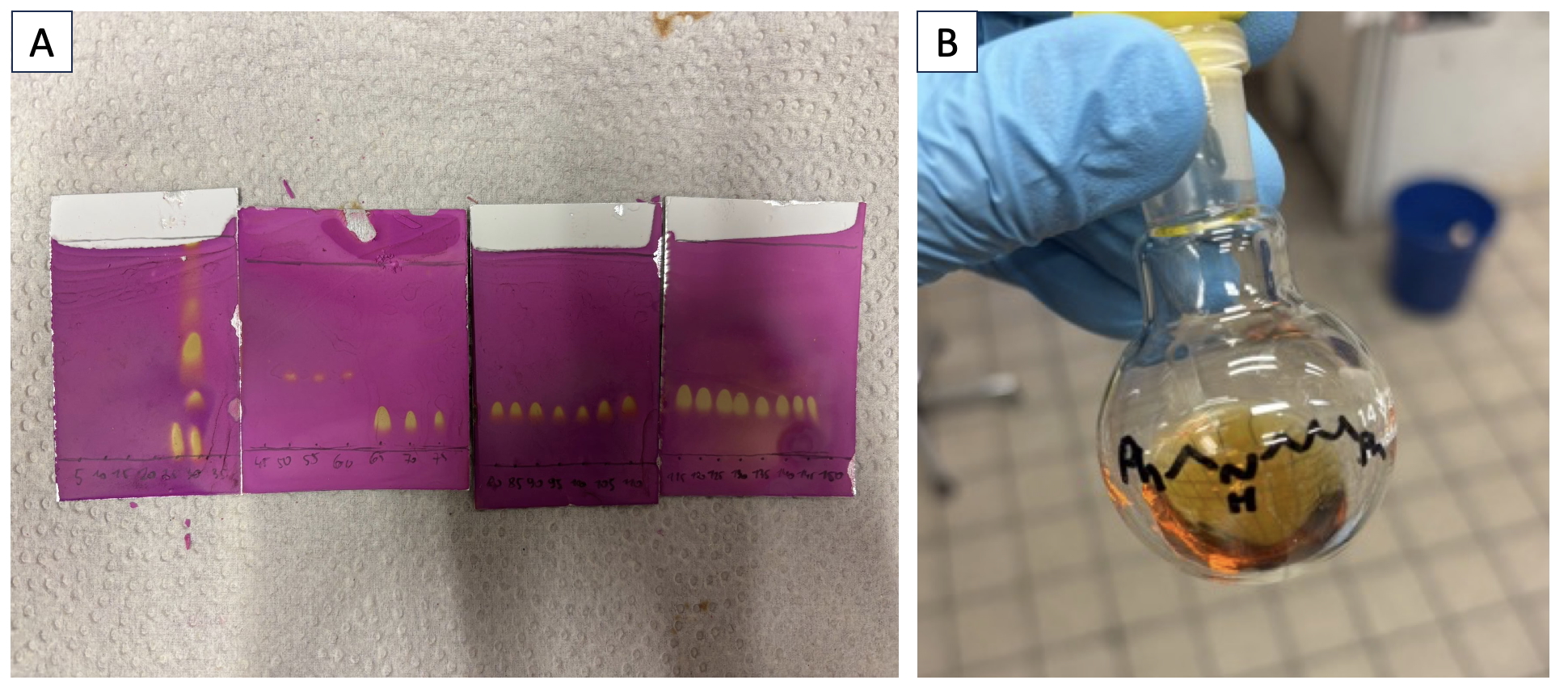
Figure 6. A. TLC analysis of collected fractions (Note 19). The product can be found in fractions 65 to 150; B. Isolated product 5.
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of “Prudent Practices in the Laboratory” (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also “Identifying and Evaluating Hazards in Research Laboratories” (American Chemical Society, 2015) which is available via the associated website “Hazard Assessment in Research Laboratories” at
https://www.acs.org/about/governance/committees/chemical-safety.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with formaldehyde,
dibenzylamine,
dichloromethane,
potassium carbonate,
4-phenyl-1-butene,
potassium hydroxide,
trifluoroacetic acid, silica gel,
ethyl acetate and
heptane.
2.
Dibenzylamine (98%) was purchased from abcr GmbH and used as received.
3. Formaldehyde (37% solution in water) was purchased from Sigma Aldrich and used as received.
4.
Ethyl acetate was purchased from Fisher Scientific and used as received.
5.
Sodium sulfate (reagent grade) was purchased from VWR Chemicals and used as received.
6. The formation of the aminal was repeated on the same scale (32.7 g, 89%).
7. Compound
3:
1H NMR
pdf (500 MHz, CDCl
3) δ 7.33 - 7.27 (m, 16H), 7.25 - 7.20 (m, 4H), 3.62 (s, 8H), 3.11 (s, 2H).
13C NMR
pdf (126 MHz, CDCl
3) δ 139.9, 129.1, 128.3, 126.9, 72.5, 56.3. FT-IR: 3082, 3061, 3025, 2925, 2880, 2835, 2795, 1603, 1494, 1451, 1406, 1365, 1244, 1123, 1073, 1028, 975, 738, 697 cm
-1. HRMS (ESI): Calculated for [M+H]
+ (C
29H
31N
2)
+: 407.24818; Found: 198.1270 (corresponding to
N,
N-dibenzylamine).
8.
Trifluoroacetic acid (99%) was purchased from abcr GmbH and used as received.
9.
4-Phenyl-1-butene (98%) was purchased from abcr GmbH and used as received.
10.
Sodium hydroxide (97%) was purchased from Macron fine chemicals and used as received.
11. 1 M aqueous solution of
NaOH was prepared by dissolving 1.6 g of
NaOH in 40 mL of distilled water.
12.
Dichloromethane was purchased from Fisher Scientific and used as received.
13. 5 M aqueous solution of
NaOH was prepared by dissolving 4 g of
NaOH in 100 mL of distilled water. Caution: exothermic!
14. For more polar amines, an eluent system of
heptane: EEA (73%
ethyl acetate, 25% ethanol, 2% of 30% aqueous solution of
NH4OH) can be used.
15. Silica gel 60 was purchased from VWR chemicals and used as received.
16. Column dimensions: 200 x 500 mm, approximately 150 g of SiO
2 used. SiO
2 was added to the column, after which the column was flushed 5 times with 600 mL of 10%
EtOAc in
heptane. Before loading the sample, a 3-cm thick layer of washed sand was added, to ensure that the bed of silica gel is not disturbed.
17.
Heptane was purchased from Sigma Aldrich and used as received.
18. TLC analysis is performed on TLC plates purchased from Merck (TLC Silica gel 60 F254). Rf = 0.2 (100%
Ethyl acetate, silica on aluminum,
KMnO4 stain).
19. A second run on the same scale gave 4.01 g (79% yield) of product
5.
20. Compound
5:
1H NMR
pdf (500 MHz, CDCl
3) δ 7.28 - 7.24 (m, 4H), 7.24 - 7.17 (m, 3H), 7.14 - 7.10 (m, 3H), 3.73 (s, 2H), 2.56 (dt,
J = 9.5, 7.4 Hz, 4H), 1.62 - 1.55 (m, 2H), 1.50 (ddd,
J = 14.9, 8.4, 7.0 Hz, 2H), 1.41 (s, 1H), 1.36 - 1.29 (m, 2H).
13C NMR
pdf (126 MHz, CDCl
3) δ 142.8, 140.6, 128.5 (2C), 128.4, 128.3, 127.0, 125.8, 54.2, 49.5, 36.0, 31.5, 30.1, 27.1. FT-IR: 3063, 3025, 2928, 2855, 2814, 1496, 1453, 1119, 730, 697 cm
-1. HRMS (ESI): Calculated for [M+H]
+ (C
18H
24N)
+: 254.19033; Found: 254.1891. Purity was assessed at 98.4% by quantitative
1H NMR
pdf spectroscopy using dibromomethane as an internal standard.
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Aliphatic amines are among the most prevalent structural moieties in organic chemistry, present in numerous natural products and pharmaceuticals.
2,3 Regardless of the ubiquity of alkylamines, the use of established synthetic methods to access and modify these motifs often results in poor yields due to limited selectivity, harsh reaction conditions, or functional group incompatibility.
3,4 Although traditional methods, such as alkylation and reductive amination,
3,5 continue to be the primary strategies for their synthesis, tremendous progress has recently been achieved in the development of transformations that facilitate direct addition of nitrogen-containing building blocks to alkenes.
3,6 In this regard, hydroaminoalkylation has emerged as a highly efficient C-C coupling reaction that facilitates the overall addition of an amine and a hydrogen across an olefin.
3, 6a, 7 While extensive studies have been conducted with both early and late transition-metal catalysts, the reported methodologies tend to deliver branched products and/or are limited to activated and conjugated alkenes.
7To address some of the persistent limitations, we reported a complementary approach for hydroaminoalkylation of unactivated alkenes and alkynes that eliminates the need for metal catalysts and external reducing agents (Scheme 1A).
8 In contrast to the previously mentioned transition-metal catalyzed methods, this reaction design, which relies on an internal redox event, enables the exclusive formation of linear products. This synthetic method employs readily available, bench-stable reagents, yielding the desired amines with high efficiency and excellent functional group tolerance. The reaction's utility was further showcased by
in situ entrapment of an iminium ion (reaction's resting state) with various nucleophiles, which led to the formation of additional functionalized tertiary amines. (Scheme 1B).
Scheme 1. A. Selected scope examples; B. Selected examples of domino synthesis of tertiary amines.
Appendix
Chemical Abstracts Nomenclature (Registry Number)
Dibromoethane; (74-95-3)
Dibenzylamine; (103-49-1)
Formaldehyde; (50-00-0)
N,N,N',N'-Tetrabenzylmethanediamine; (3) (58288-30-5)
Trifluoroacetic acid; (76-05-1)
4-Phenyl-1-butene; (768-56-9)

|
Uroš Vezonik obtained an MSc in Chemistry from the University of Ljubljana, where he conducted research under the supervision of Prof. Janez Košmrlj. During his master's studies, he completed a research visit at the University of Illinois Urbana-Champaign with Prof. David šarlah, focusing on the total synthesis of marine triterpenoids. Since 2022, he has been a graduate student in the group of Prof. Nuno Maulide, where he is developing novel synthetic methodologies based on highly reactive electrophilic species, with applications in the synthesis of natural products and pharmaceutically relevant compounds. |

|
Paolo Piacentini was born in Alessandria, Italy, in 1995. He earned his Ph.D. in 2023 from Pavia University under the supervision of Prof. David Sarlah. In 2022, he joined Prof. Nuno Maulide's research group as a postdoctoral fellow, focusing on entropy-oriented drug design. Currently, he is a postdoctoral researcher at Merck KGaA, Germany, specializing in Medicinal Chemistry and Automation. |

|
After obtaining his Ph.D. at the Université catholique de Louvain under the supervision of Prof. István E. Markó in 2007 Nuno Maulide pursued a postdoctoral position in the group of Prof. Barry M. Trost at Stanford University. In 2009, he started his independent career at the Max-Planck Institut für Kohlenforschung and only 4 years later, in 2013 he became a Full Professor at the University of Vienna. His research interests are broadly spread in the field of organic chemistry and include the development of new reaction methods, the total synthesis of natural compounds and medicinal chemistry driven investigations. He is a member of the Board of Editors for Organic Syntheses (2018). |

|
Anagha Veluthanath Nair obtained her integrated BS/MS degree in Chemical sciences from the Indian Institute of Science Education and Research, Thiruvananthapuram, India, in 2023. During her master's, she worked with Dr. Basudev Sahoo on Cu(I) catalyzed alkyl borylation of strained alkenes and metal-photoredox catalysis for N-heterocycle functionalization. Since 2023, she has been a graduate student in the group of Prof. M Kevin Brown, where she is developing novel synthetic methodologies utilizing deaminative coupling of primary N-Nitroso carbamates with various electrophiles. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved