Org. Synth. 2025, 102, 428-441
DOI: 10.15227/orgsyn.102.0428
Hydrosilylation of Propiolate Esters
Submitted by Allison M. Clark, Giulianna Miseo, and Neil K. Garg*
1Checked by Seamus Higgins and Christopher Vanderwal
1. Procedure (Note 1)
A. Silyl acrylate (2). A single-necked (24/40 joint) 250-mL round-bottomed flask is equipped with a Teflon-coated magnetic stir bar (football-shaped, 2.5 x 1.5 cm) and fitted with a 24/40 rubber septum. The apparatus is flame-dried under reduced pressure and allowed to cool to 23 ℃ under a nitrogen atmosphere. The flask is then charged with AlCl3 (7.5 g, 56 mmol, 1.1 equiv) (Figure 1A) by quickly removing the septum, adding the solid, and then placing the flask back under a nitrogen atmosphere (Note 2). The flask is maintained under an atmosphere of nitrogen during the course of the reaction. To the flask is then added CH2Cl2 (51 mL, 1.0 M) (Note 3) via syringe and stirring (700 rpm) is commenced, leading to a cloudy yellow solution (Figure 1B). The flask is placed in an ice bath and the mixture is allowed to cool to 0 ℃ over 5 min. Then, Et3SiH (8.1 mL, 51 mmol, 1.0 equiv) (Note 4), is added dropwise over 5 min via syringe, generating a white and opaque suspension (Figure 1C). Next, ethyl propiolate (5.2 mL, 51 mmol, 1.0 equiv) (Note 5) is added via a syringe dropwise over 5 min, generating first a light yellow, foamy solution, which then turns into a dark yellow, clear solution by the completion of the addition (Figure 1D). Following addition of all reagents, the reaction mixture was stirred at 700 rpm for 2 h at 0 ℃ (Note 6).

Figure 1: A. Reaction flask with AlCl3; B. Cooling flask after addition of CH2Cl2; C. Flask after addition of Et3SiH; D. Flask after addition of ethyl propiolate; E. Reaction mixture after 2 h at 0 ℃
After the allotted reaction time, the mixture appears as a dark yellow, clear solution (Figure 1E). The 24/40 rubber septum is removed. With the reaction mixture still in the 0 ℃ bath, saturated aqueous NaHCO3(10 mL) (Note 7) is added dropwise over 10 min, then an additional 140 mL is added in 5 mL portions over 35 min to quench the AlCl3. The reaction flask is then taken out of the ice bath and allowed to warm to 23 ℃ over 30 min (Note 8). The biphasic reaction mixture is transferred to a 500-mL separatory funnel and the round-bottomed flask is rinsed once with CH2Cl2 (50 mL). Then, brine (100 mL) (Note 9) is added and the biphasic mixture is shaken vigorously with intermittent venting. The layers are separated (Figure 2A), and the organic layer (CH2Cl2) is collected in a 500-mL Erlenmeyer flask (Note 10). The aqueous layer is extracted twice more with CH2Cl2 (50 mL). The pooled organic layers from the three extractions are combined and transferred into the 500-mL separatory funnel. Then, brine (150 mL) is added and the separatory funnel is vigorously shaken. The organic layer is collected in another 500-mL Erlenmeyer flask and dried over sodium sulfate (50 g) (Na2SO4) (Note 11). Then, the solid Na2SO4 is removed by vacuum filtration through a Büchner funnel equipped with filter paper (inner diameter: 8.0 cm), and the filtrate is collected into a 500-mL Erlenmeyer flask (Figure 2B). The solid Na2SO4 is subsequently rinsed with CH2Cl2 (50 mL x 2). The collected solution is then transferred into a 500-mL round-bottomed flask and concentrated by rotary evaporation (200 mbar, 30 ℃ bath temperature), then placed under reduced pressure (0.630 Torr) for 1 h to afford the crude product as a clear, light-yellow oil (Note 12).

Figure 2. A. Separation of organic and aqueous layers during extraction; B. Vacuum filtration apparatus
The material is then transferred to a single-necked (14/20) 100-mL pear-shaped round-bottomed flask equipped with a magnetic stir bar (football-shaped, 1.6 x 0.75 cm). This flask is fitted with a short path distillation apparatus and a tared, pear-shaped single-necked (14/20) 50-mL round-bottomed flask equipped with a tared stir bar (football-shaped, 1.6 x 0.75 cm) is secured with a Keck clip (14/20) to the collection arm. The short path distillation apparatus is equipped with a thermometer, and the flask is submerged in an oil bath (Figure 3A). Stirring is commenced (700 rpm) and the bath is heated gradually from 23 to 90 ℃ over a period of 1 h at 0.630 Torr (Note 13). The product and triethylsilane are collected as a mixture by distillation at 40-55 ℃ (0.630 Torr) into the tared 50-mL pear-shaped round-bottomed flask. The distillate appears as a clear liquid, leaving behind a brown residue (Figure 3B). After the distillation, the collection flask with the product mixture is placed under reduced pressure (0.630 Torr) in an oil bath at 40 ℃ with stirring (700 rpm) for 6 h (Note 14) to remove remaining triethylsilane (Figure 3C). The desired product is obtained as a clear liquid (6.97 g, 63% yield, 98.5% purity) (Notes 15 and 16).

Figure 3. A. Distillation setup; B. Completed distillation; C. Heating final product under reduced pressure
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of “Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also “Identifying and Evaluating Hazards in Research Laboratories” (American Chemical Society, 2015) which is available via the associated website “Hazard Assessment in Research Laboratories” at
https://www.acs.org/about/governance/committees/chemical-safety.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with vacuum distillation,
ethyl propiolate,
AlCl3,
Et3SiH, hexanes,
ethyl acetate, deionized
H2O,
KMnO4,
NaHCO3,
CH2Cl2,
NaCl,
Na2SO4, and
CDCl3.
2. The authors used
AlCl3 (anhydrous, 99.999% metals basis) purchased from Fisher-Scientific, which was used as received. The mass of
AlCl3 added was determined by weighing the solid directly into the flame-dried, pre-tared reaction flask in a fume hood. Before removing it from the fume hood to bring to the balance, the flask was sealed with a pre-weighed septum. The checkers used
AlCl3 (anhydrous, 98.5% metals basis) purchased from Alfa-Aesar and observed no discernible differences.
3. The authors used
methylene chloride (99.9%) purchased from Fisher Chemical. It was passed through an activated alumina column before use.
4. The authors used
Et3SiH (99%) purchased from Sigma-Aldrich Corp., which was used as received.
5. The authors used
ethyl propiolate (99%) purchased from Sigma-Aldrich Corp., which was used as received.
6. The progress of the reaction is monitored by TLC analysis using a diluted aliquot of the reaction mixture. This is prepared by placing one drop of the reaction mixture solution (using a 9-inch Pasteur pipette) into a 13 x 100 mm test tube and diluting with 1 mL of
CH2Cl2. This solution is spotted on silica gel (Figure 4) and 9:1 Hexanes:
ethyl acetate is used to elute. The plate is visualized using a
potassium permanganate (
KMnO4) stain and a 254 nm UV lamp.
Ethyl propiolate appears at Rf: 0.44 (UV inactive, yellow spot) and the silylated product appears at Rf: 0.59 (UV active, yellow spot). The authors used
ethyl acetate (99.5%) and
n-hexanes (98.5%) from Fisher Scientific, which were both used as received.
Figure 4. TLC of reaction after 2 h, developed using 9:1 Hex:EtOAc (SM = Ethyl propiolate, CO = Co-spot, RXN = Diluted reaction mixture)
7.
NaHCO3 must be cautiously added one drop at a time, or the reaction could overflow. The authors used
NaHCO3 purchased from J. T. Baker. Saturated aqueous
NaHCO3 solution is prepared by dissolving solid
NaHCO3 (250 g) in deionized water (2000 mL).
8. The flask contents should not be transferred to the separatory funnel until the bubbling has ceased and the mixture has warmed to 23 ℃.
9. The authors used
NaCl (>99.5%) purchased from VWR. Brine solution is prepared by dissolving solid
NaCl (250 g) in deionized water (2000 mL).
10. An emulsion forms at the interface of the organic and aqueous layer that can be reduced by swirling the flask and letting the mixture sit for 5-10 min. If the indicated volume of brine is inadequate to separate the layers, an additional 50-100 mL of brine may be added.
11. The authors used
Na2SO4 (>99%) purchased from Fisher Scientific, which was used as received.
12. The checkers concentrated the collected solution by rotary evaporation (50 mm Hg vacuum strength, 30 ℃ bath temperature), then placed under reduced pressure (0.380 Torr) for 1 h and observed no discernible differences.
13. The vacuum line on the distillation head was connected directly to a Schlenk line, which was measured to pull vacuum at 0.630 Torr (Figure 5). The temperature at which the product distills at may differ if the vacuum strength is different. In replicating this procedure, the checkers used the described distillation set up which was measured to pull vacuum at 0.380 Torr.
Figure 5. Vacuum strength gauge for distillation
14. At this point, the product is nearly pure with only minor amounts of
triethylsilane remaining. Heating under reduced pressure completely removes this impurity, however some of the product may be lost in this process. In duplicating this procedure, the authors only heated the product under vacuum for 3 h. The checkers found that heating under reduced pressure (0.380 Torr) for 3 h produced satisfactory results.
15. The purity of the product was determined to be approximately 97.9% by qNMR
pdf using
1,3,5-trimethoxybenzene (Alfa Aesar, 99%) as the internal standard. The checkers found the purity of the product to be >97% by qNMR using
1,3,5-trimethoxybenzene (Sigma Aldrich, 99%) across three runs.
16. The purified product acrylate
2 is characterized as follows:
1H NMR
pdf (400 MHz, 298 K,
CDCl3) δ 6.83 (d,
J = 3.0 Hz, 1H), 5.97 (d,
J = 3.0 Hz, 1H), 4.20 (q,
J = 7.1 Hz, 2H), 1.30 (t,
J = 7.1 Hz, 3H), 0.93 (t,
J = 7.9 Hz, 9H), 0.71 (q,
J = 8.6, 6H).
13C NMR
pdf (101 MHz, 298 K,
CDCl3) δ 169.71, 141.56, 140.18, 60.36, 14.19, 7.27, 3.03. IR
pdf (film): 2954.24 cm
-1, 2875.55 cm
-1, 1718.97 cm
-1, 1703.06 cm
-1, 1463.16 cm
-1. HRMS-ESI (
m/z) [M + Na]
+ calc'd for C
11H
22O
2SiNa
+, 237.1287; found, 237.1291; Rf 0.59 (9:1 hexanes:
EtOAc). The authors determined the boiling point to be 85 ℃ at 0.630 Torr. The checkers determined the boiling point to be 76 ℃ at 0.380 Torr.
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
The procedure described provides an efficient, scalable, and chromatography-free procedure for the regioselective hydrosilylation of propiolate esters, employing a Lewis acid to impart regioselectivity. Methods to install silicon functional groups are important as silanes are valuable moieties in organic and materials chemistry. Alkyl, alkoxy, amino, or mercapto silanes have been applied to scavenge moisture and provide stabilizing protection to a variety of materials, including plastics, dental materials, and mineral fillers. They can also been used to improve adhesive properties in paints, inks, or sealants.
2Hydrosilylation reactions have been widely applied in polymerizations or for post-polymerization functionalization of polysiloxanes. For example, it is a highly popular method for chain end modification of polydimethylsiloxane (PDMS), such as by hydrosilylation of allyl alcohol to yield α,ω-dihydroxypropyl PDMS
3 (Figure 6).
3,4 These functionalized polymers can be carried forward to telechelic silicone oligomers that serve as macromonomers in numerous step-growth, free-radical, and ring-opening polymerization reactions.
5Furthermore, vinyl silanes are valuable to the pharmaceutical industry due to their unique properties as carbon bioisosteres.
6 Specifically, Si-H bonds exhibit stronger hydrogen bonding than C-H bonds due to increased bond length, and silyl groups in small molecules can also offer increased lipophilicity and improved ADME properties. Consequently, many biologically active molecules containing silicon have been recognized. For example, compound
4 is a CBD analog with anti-inflammatory properties against NLRP3 inflammasome, which could be useful in treating hemolytic diseases such as sickle cell anemia (Figure 6).
7Organosilanes are not only used in industrially relevant products but are also highly valuable to organic chemists as versatile synthetic intermediates due to their steric and electronic properties that enable unique modes of reactivity. The specific types of products formed in our procedure herein (i.e.,
5, Figure 6) are especially useful. For example, Padwa and coworkers demonstrated how the bulkiness of the α-silyl group in
5 can induce complementary regioselectivity to its H-analog, as in the [3+2] cycloaddition between
5 and nitrile oxide
6 to give isoxazoline
7 with loss of the silyl group (Figure 6).
8 Additionally, the α-silyl group of compound
5 can affect an unusual 1,4 selectivity in the nucleophilic addition of organolithium and Grignard reagents to the acrylate, rather than 1,2-addition.
9 This allows direct formation of a C-C bond at the β position of the acrylate, a transformation that would otherwise require the use of an organocuprate. Moreover, the remaining silyl functional group in the product
8, may be leveraged to enable further transformations, such as in the desilylative reaction of α-silyl carboxylic acid
8 with aryl trifluoromethyl ketone
9 to form β-lactone
10.
10 Finally, the α-silyl group of acrylate
5 can enable nucleophilic reactivity at the α-carbon of an acrylate. For example, fluoride-mediated addition into aldehyde
11 furnishes unsaturated β-hydroxy ester
12.
11
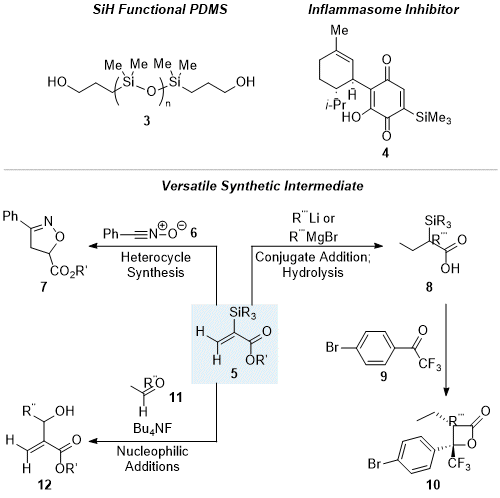
Figure 6. Examples of organosilanes and applications of silanes 5
Generally, methods for organosilane synthesis include transition metal catalysis, nucleophilic addition of silyl-metal reagents, and addition of carbon nucleophiles (R-Li, R-MgBr, etc.) to silyl halides.
12 The hydrosilylation of propiolate esters
13 detailed herein provides an alternative, atom-economical strategy towards production of synthetically versatile acrylates
5. This methodology was developed by Yamabe and coworkers and offers versatility in silicon electrophile choice, acrylate choice, and tunable regioselectivity.
13 The initial study demonstrated that this method can be used to install either triethylsilyl or tris(trimethylsilyl)silyl groups, as shown in products
2 and
14, respectively (Figure 7). Both silyl groups could also be added to trifluoroethyl propiolate esters, yielding triethylsilyl-substituted acrylate
15 and tris(trimethylsilyl)silyl substituted acrylate
16.
Figure 7. Select scope of hydrosilylations of propiolate esters
Yamabe and coworkers proposed a viable mechanism for this hydrosilylation (Figure 8). Hydride addition occurs on the electrophilic β-carbon of 1 bound to AlCl3. Subsequent nucleophilic addition of the resulting allenolate 17 to tris(trimethylsilyl)silylium cation leads to the formation of silyl acrylate 14.
Figure 8. Proposed mechanism of hydrosilylation reaction
Overall, this procedure provides a simple approach for accessing α-silyl acrylates. The low cost of the starting materials, ease of setup, excellent regioselectivity and chromatography-free purification process make it an attractive procedure. Furthermore, the silyl acrylate esters formed serve as versatile substrates for the preparation of value-added products.
14
Appendix
Chemical Abstracts Nomenclature (Registry Number)
AlCl3; (7446-70-0)
Et3SiH; (617-86-7)
ethyl propiolate; (623-47-2)

|
Allison Clark was born in Los Altos, CA, and received her B.S. in Chemistry from the University of Southern California, where she carried out research under the direction of Professor Megan Fieser. In 2023, she then moved to the University of California, Los Angeles, where she is currently a third-year graduate student in Professor Neil K. Garg's Laboratory. Her studies primarily focus on developing synthetic methods utilizing strained intermediates. |

|
Giulianna Miseo was born Livingston, NJ and raised in Bernardsville, NJ. In 2020, she received her B.S. in Biochemistry/Chemistry from the University of California, San Diego. In 2023, she began her graduate studies at the University of California, Los Angeles, where she is currently a third-year graduate student in Professor Neil K. Garg's Laboratory. Her studies are primarily focused on total synthesis. |

|
Neil Garg is the Distinguished Kenneth N. Trueblood Professor of Chemistry at the University of California, Los Angeles. His laboratory develops novel synthetic strategies and methodologies to enable the total synthesis of complex bioactive molecules. |

|
Seamus Higgins earned his B.S. in Chemistry from the University of Vermont in 2023, where he studied 1,3 diaza-Claisen rearrangements with Prof. Jose S. Madalengoitia. He is currently a graduate student in the lab of Prof. Christopher D. Vanderwal at the University of California, Irvine, working in the areas of total synthesis and medicinal chemistry. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved