Org. Synth. 2025, 102, 465-476
DOI: 10.15227/orgsyn.102.0465
Oxidative Nitrogen Atom Insertion into Cyclopentenones
Submitted by Bence B. Botlik and Bill Morandi*
1Checked by Maud T. H. Tregear, Anna J. Passmore, and Darren J. Dixon
1. Procedure (Note 1)
7-Methoxyisoquinolin-1(2H)-one (2). In air, a 1-L one-necked round-bottomed flask (29/32) equipped with a Teflon-coated egg-shaped magnetic stir bar (40 x 20 mm) was consecutively charged with 6-methoxy-2,3-dihydro-1H-inden-1-one (4.86 g, 30.0 mmol, 1.0 equiv) (Note 2), tetrahydrofuran (THF, 210 mL, 0.14 M) (Note 2), triethylamine (Et3N, 4.5 g, 6.3 mL, 45 mmol, 1.5 equiv, added via syringe) (Note 2) and tert-butyldimethylsilyl trifluoromethanesulfonate (TBSOTf, 9.51 g, 8.25 mL, 36 mmol, 1.2 equiv, added via syringe) (Notes 2 and 3), in this order of addition, while constant stirring was applied (Note 4). The resulting mixture was stirred at room temperature (Notes 4 and 5) for 1 h, and then methanol (MeOH, 210 mL) (Note 2) was added into the flask in one portion. The resulting mixture was stirred at room temperature for 5 min (Note 6), and then ammonium carbamate (H2NCO2NH4, 9.36 g, 120 mmol, 4.0 equiv) (Notes 2 and 7) was added into the flask in one portion. The resulting mixture was stirred at room temperature for 5 min (Note 6). (Diacetoxyiodo)benzene (PIDA, 29.0 g, 90 mmol, 3.0 equiv) (Notes 2, 8 and 9) was then added into the flask in one portion at room temperature, and the resulting mixture was stirred at room temperature for 30 min (Notes 6 and 10). The magnetic stir bar was then removed, and the mixture was filtered through a filter paper into a 1-L one-necked round-bottomed flask (Note 11), and it was concentrated by rotary evaporation (Note 12). The resulting crude mixture was dissolved in dichloromethane (DCM, 200 mL) (Note 2), transferred into a 1-L separatory funnel (NS 29, Teflon stopper), and the mixture was thoroughly shaken with saturated aqueous sodium hydrogen carbonate solution (NaHCO3, 250 mL) (Note 2), until the initial gas evolution stopped. The aqueous phase was subsequently washed with dichloromethane two times (2 x 200 mL DCM). The organic phases were combined and washed with brine (200 mL), before being dried by stirring over sodium sulfate (Na2SO4, 150 g) (Note 2) for 10 min and filtered through a filter paper (Note 11) into a 1-L one-necked round-bottomed flask. Silica gel (30 g) (Note 13) was added to the solution, and the mixture was concentrated by rotary evaporation (Note 14). The crude reaction mixture was purified by flash column chromatography, using DCM:MeOH = 100:0 to 95:5 eluent (Note 15). The fractions containing the product were combined into a 1-L one-necked round-bottomed flask and concentrated by rotary evaporation (Note 16). The product was then dried under high vacuum (Note 17). No further purification was required, affording 3.55 g of 7-methoxyisoquinolin-1(2H)-one (2) as a fine beige powder in 68% yield and 100% purity (Notes 18, 19, 20, and 21).

Figure 1. Reaction set-up. A. The reaction mixture after the addition of TBSOTf; B. The reaction mixture 30 min after the addition of PIDA. (Photos provided by the authors)
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of “Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also “Identifying and Evaluating Hazards in Research Laboratories” (American Chemical Society, 2015) which is available via the associated website “Hazard Assessment in Research Laboratories” at
https://www.acs.org/about/governance/committees/chemical-safety.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
6-methoxy-2,3-dihydro-1H-inden-1-one,
tetrahydrofuran,
triethylamine,
tert-butyldimethylsilyl trifluoromethanesulfonate,
methanol,
ammonium carbamate,
(diacetoxyiodo)benzene,
dichloromethane,
sodium hydrogen carbonate,
sodium sulfate, and silica gel as well as the proper procedures for setting up experimental operations. Additional caution should be taken with regard to highly corrosive
tert-butyldimethylsilyl trifluoromethanesulfonate, and the use of
tetrahydrofuran which is prone to form peroxides.
2.
6-methoxy-2,3-dihydro-1H-inden-1-one (
1, 98%) was obtained from Fluorochem Ltd.
Tetrahydrofuran (>99.5%, stabilized with 250 ppm BHT) was obtained from Sigma Aldrich.
Triethylamine (>99%) was obtained from Sigma Aldrich.
tert-Butyldimethylsilyl trifluoromethanesulfonate (97%) was obtained from Fluorochem Ltd.
Methanol (>99.8%) was obtained from Sigma Aldrich.
Ammonium carbamate (>97%) was obtained from Tokyo Chemical Industry Co., Ltd.
(Diacetoxyiodo)benzene (98%) was obtained from Fluorochem Ltd.
Dichloromethane (>99%, stabilized by amylene) was obtained from Sigma Aldrich.
Sodium hydrogen carbonate (>99.7%) was obtained from Fisher Scientific.
Sodium sulfate (>99.5%) was obtained from Fisher Scientific.
1,3,5-Trimethoxybenzene was obtained from Fluorochem. All chemicals were used as received.
3. Upon the addition of
tert-butyldimethylsilyl trifluoromethanesulfonate, fumes appear above the reaction mixture, which gradually fade away.
4. A MR Hei-Tec stirring plate obtained from Heidolph Instruments GmbH & Co. was used throughout this manuscript. A stir rate of 400 rpm was applied in this step.
5. Room temperature throughout this manuscript refers to temperature between 20 ℃ and 25 ℃.
6. A stir rate of 500 rpm was applied in this step.
7. 4 equivalents of
ammonium carbamate are used in the reaction in order to achieve the highest possible yields. The use of less equivalents of
ammonium carbamate results in lower, but still synthetically useful yields. The addition of
ammonium carbamate containing larger lumps can result in a heterogeneous mixture, however, this does not affect the outcome of the reaction.
8. Upon the addition of
(diacetoxyiodo)benzene, the reaction mixture turns yellow (Figure 1B).
9. Due to the oxidative nature of the reaction (4-electron oxidation), 2 equivalents of
(diacetoxyiodo)benzene are needed in the reaction by stoichiometry. 3 equivalents of
(diacetoxyiodo)benzene are utilized in order to achieve the highest possible yields, which corresponds to 50% excess with regard to the theoretically required amount.
10. The reaction can be monitored by TLC (
SiO2,
DCM:
MeOH = 95:5 eluent, starting material
1: R
f = 0.83, product
2: R
f = 0.28; visualization with
KMnO4, Figure 2). SM refers to starting material
1, RM refers to the final reaction mixture after the workup, P refers to product
2.
Figure 2. TLC monitoring of the reaction (Photo provided by the authors)
11. A 150 mm diameter, MN 615 grade filter paper obtained from Macherey-Nagel was used in the filtration steps. The purpose of the first filtration step is to remove a small number of undissolved
ammonium carbamate particles, this can be skipped if grounded
ammonium carbamate is used, containing only fine powder, without the presence of larger lumps.
12. Throughout this procedure, a rotary evaporator R-300 purchased from BÜCHI Labortechnik AG was used, with a rotation rate of 240 rpm, and the temperature of the water bath was set to 40 ℃. The use of elevated temperature has no safety risk, as the oxidant is fully consumed in the reaction. To remove the
tetrahydrofuran-
methanol solvent mixture, the following drying method was used: 40 min at 180 mm Hg, 30 min at 135 mm Hg, 5 min at 75 mm Hg, 10 min at 18 mm Hg.
13. Silica gel (230-400 mesh) was obtained from Sigma Aldrich and used as received.
14. To remove the
dichloromethane solvent, the following drying method was used: 25 min at 450 mm Hg, 5 min at 375 mm Hg, 5 min at 300 mm Hg, 10 min at 18 mm Hg.
15. A column was wet-packed with a slurry of 200 g of silica gel (
Note 13) in 0.5 L of
DCM, resulting in a column of silica slurry with 7 cm diameter and 18 cm height (Figure 3). The reaction mixture distributed on silica was dry-loaded onto the column through a powder funnel, and subsequently 55 g sand was layered onto the column. The column was flushed with 750 mL
DCM, followed by 500 mL
DCM:
MeOH 99.5:0.5, then 500 mL
DCM:
MeOH 99:1, then 500 mL
DCM:
MeOH 98:2, and finally 250 mL
DCM 97:3 eluent. Fraction collection (50-mL fractions) was started, and elution was continued with 250 mL
DCM:
MeOH 97:3 eluent. The eluent was then changed to 500 mL
DCM:
MeOH 96:4 system, and finally 1 L
DCM:
MeOH 95:5. 41 fractions were collected in total, among which fractions 12-40 contained the desired product, and a minor co-eluting impurity (observed by TLC,
SiO2,
DCM:
MeOH = 95:5 eluent, (R
f of product
2 = 0.28, visualization with
KMnO4). Only fractions 19-40 were collected to avoid this impurity, and fractions 12-18 were subject to additional purification. With the second column, 22 fractions were collected, of which fractions 3-16 contained the desired product and a minor impurity. Only fractions 7-16 were collected due to this impurity.

Figure 3. Flash column chromatography of the reaction mixture (photo provided by the checkers)
16. To remove the
dichloromethane-
methanol solvent mixture, the following drying method was used: 25 min at 525 mm Hg. The product was then transferred into a 500-mL one-necked round-bottomed flask, then 15 min at 525 mm Hg, 5 min at 375 mm Hg, 10 min at 18 mm Hg.
17. High vacuum was established with a vacuum oil pump provided by Vacuubrand GmbH & Co. KG. For drying, room temperature and a pressure lower than 1.5 mm Hg were maintained for 1 h.
18. Purity was determined by qNMR
pdf, using
1,3,5-trimethoxybenzene internal standard in
d6-DMSO.
19. Characterization of product
2: Appearance: beige fine powder (
Figure 4)
1H NMR
pdf (400 MHz, DMSO) δ 11.20 (s, 1H), 7.63 - 7.56 (m, 2H), 7.31 (dd,
J = 8.7, 2.8 Hz, 1H), 7.08 - 7.01 (m, 1H), 6.51 (d,
J = 7.1 Hz, 1H), 3.85 (s, 3H).
13C NMR
pdf (101 MHz, DMSO) δ 161.5, 157.9, 131.8, 128.0, 127.3, 126.4, 122.1, 107.1, 104.5, 55.3. Melting point: 208 - 211 ℃. IR (film): 3156, 3005, 2934, 2869, 1658, 1638, 1617, 1554, 1505, 1487, 1364, 1271, 1211, 1025, 829, 787, 696 cm
-1 HRMS (ESI) m/z: [M+Na]
+ calculated for (C
10H
9NO
2Na) 198.0526; found 198.0525.
20. A second run on 15 mmol scale yielded 1.63 g of desired product in 62% yield and 100% purity. For this run, only one column was required for purification, with an eluent gradient of
DCM:
MeOH 100:0 to
DCM:
MeOH 98:2. The column was first flushed with 2.4 L
DCM, then 600 mL
DCM:
MeOH 99.5:0.5, and finally 600 mL
DCM:
MeOH 99:1. Fraction collection (50 mL fractions) was started and the eluent was changed to 1.5 L
DCM:
MeOH 98:2. 30 fractions were collected in total, of which fractions 11-27 contained the desired product. Only fractions 15-27 were collected due to a small impurity.
Figure 4. The purified product (photo provided by the checkers)
21. The authors utilized recrystallization to further purify the product following flash chromatography. The solid material was dissolved in boiling
DCM while maintaining intensive stirring with a stirring plate and a stirring bar (gradually increasing the stirring rate as the amount of the added solvent increased). In total, 300 mL
DCM was added, and due to evaporation, 280 mL solution remained at the point when all product dissolved (the stirring lasted approximately 1.5 h). The mixture was left to cool down to room temperature in approximately 3 hours, during which the flask was open to air and was under fume hood ventilation. The mixture was then capped (NS 29, Teflon stopper) and stored in a fridge (temperature = 10 ℃) for 16 hours, by the end of which time white crystals formed at the bottom of the flask. The mixture was then transferred into a freezer (temperature = -30 ℃) where it was stored for 24 hours, after which time an off-white solid formed, floating on the top of the mother liquor. The solid was rapidly filtered using a sintered funnel over vacuum, while the mixture was still cold. The solid product, distributed on the filter was dried by the suction of air through the filter for 5 min.
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Pyridones are prevalent moieties in natural products, drug molecules, and are commonly used as ligands in Pd-catalysed C-H activation reactions, therefore they have high importance in synthetic organic chemistry.
2,3The common methods for the synthesis of 2-pyridones are classical condensation reactions. The most important examples include the Guareschi synthesis, Knoevenagel-type reactions, and rearrangements of pyridine-
N-oxides or other pyridinium salts.
4,5 However, most of the traditional syntheses of 2-pyridones suffer from significant limitations, namely the need to synthesise the functionalised starting materials, the use of harsh reaction conditions, or not general.
In contrast to these methods, we hypothesised that a different strategy, skeletal editing, could provide easy access to 2-pyridones. We envisaged that the oxidative nitrogen atom insertion into commercially available cyclopentenones would allow us to achieve the desired transformation. It was found that the silyl enol ethers that cyclopentenones can generate
in situ in the presence of a silylating agent, can undergo nitrogen atom insertion upon reaction with (diacetoxyiodo)benzene and ammonium carbamate, and subsequent aromatisation provides 2-pyridone products.
6The reaction exhibits broad substitution pattern- and functional group tolerance. Unsubstituted, 3-substituted, and 3,4-disubstituted pyridones are accessible through the reaction. 1-Indanone derivatives are also compatible substrates, and many substitution patterns on their skeleton was tolerated, allowing us to access densely substituted pyridone derivatives. Many functional groups, including halogens, terminal alkynes, hydroxy groups, ethers, esters and amides were all found to be tolerated in the reaction. The developed methodology also provides a facile synthetic route to
15N-labelled pyridones.
6
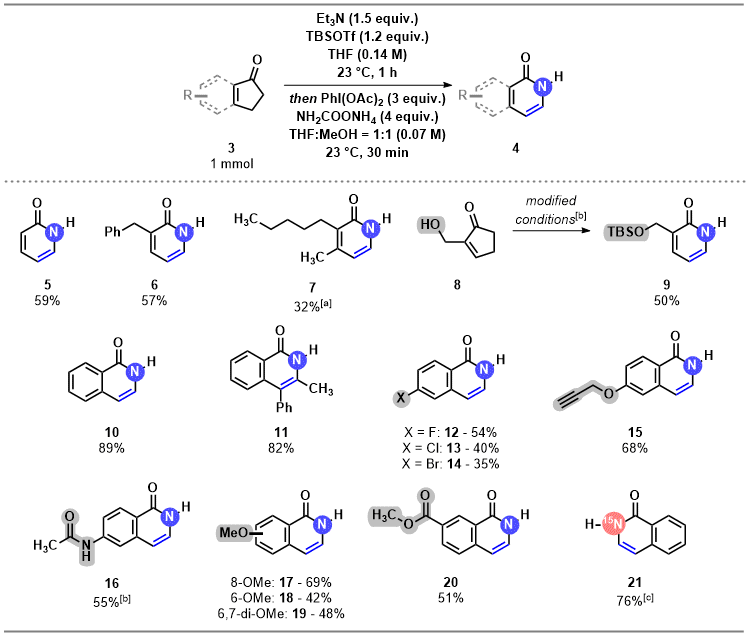
Scheme 1. Selected examples from the scope of the transformation. The yields are isolated yields. [a] Pentane used as solvent in the first step instead of THF, and pentane:methanol = 1:1 mixture as solvent in the second step. [b] 2.5 equiv of Et3N and 2.2 equiv TBSOTf were used in the first step. [c] The combination of 4.0 equiv 15NH4Cl and 4.0 equiv K2CO3 was used instead of ammonium carbamate
Appendix
Chemical Abstracts Nomenclature (Registry Number)
7-Methoxyisoquinolin-1(2H)-one; (2) (16027-16-0)
6-Methoxy-2,3-dihydro-1H-inden-1-one; (1) (13623-25-1)
Triethylamine; (121-44-8)
tert-Butyldimethylsilyl trifluoromethanesulfonate; (69739-34-0)
Ammonium carbamate; (1111-78-0)
PIDA: (Diacetoxyiodo)benzene; (3240-34-4)

|
Bence B. Botlik was born in Hungary, where he completed research internships in the groups of Prof. Zoltán Novák, Prof. Tibor Soós, and Dr. Gábor London. He earned his Master of Chemistry degree at the University of Oxford, Merton College, and during his studies he was awarded the Gibbs Trust Prize and the Brian Bannister Prize. He completed his Master thesis in the group of Prof. Véronique Gouverneur. In 2022 he started his doctoral studies in the group of Prof. Bill Morandi at ETH Zürich, where he obtained a fellowship from the Scholarship Fund of the Swiss Chemical Industry. |

|
Professor Bill Morandi studied at ETH Zürich (2003-2008), receiving a BSc in Biology and a MSc in Chemical Biology. From 2008 to 2012, he pursued his PhD in organic synthesis at the same institution in the labs of Professor Erick M. Carreira. Afterwards, he moved to the California Institute of Technology (Pasadena, CA) for a postdoctoral stay with Professor Robert H. Grubbs. From 2014 to 2018, he was an independent Max Planck Research Group Leader at the Max-Planck-Institut für Kohlenforschung (Mülheim, Germany), before subsequently returning to ETH Zürich as a Professor in 2018. He is currently Full Professor of Synthetic Organic Chemistry (Laboratorium für Organische Chemie) at ETH Zürich. |

|
Maud T. H. Tregear was born in London in 2001. She obtained her Master's in Chemistry from the University of Oxford, New College, in 2024, incorporating research internships in the groups of Sir Peter Bruce and Prof. Jonathan Morris and at AstraZeneca. During her studies, she was awarded the Part 1A Prize, Gibbs Trust Prize, Brian Bannister Prize and Salters' Graduate Award. She completed her Master's project in the group of Prof. Angela Russell. In 2024, she started her DPhil under the supervision of Prof. Darren Dixon, with her research focused on iminophosphorane reagents. |

|
Anna J. Passmore was born in Birmingham, UK in 1999. She obtained her MChem (Hons) Chemistry with industrial experience degree from the University of Manchester in 2023, completing an industrial placement at Lubrizol in her third year. Her final year project was carried out under the supervision of Dr Giacomo Crisenza, and during her studies she was awarded the Roger Grice Prize and an Outstanding Academic Achievement Award. She is now a second-year DPhil student in the group of Prof. Darren J. Dixon at the University of Oxford, working on iridium-catalyzed reactions, where she has been awarded a fellowship from the Royal Commission for the Exhibition of 1851. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved