Org. Synth. 2026, 103, 4-18
DOI: 10.15227/orgsyn.103.0004
Iridium-Catalyzed Borylation of Silanes
Submitted by Christina A. Rivera, Benjamin A. Janda, and Neil K. Garg*
1 Checked by James W. Pearson and Sophie A. L. Rousseaux*
21. Procedure (Note 1)
A. Triethyl(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)silane (3). A two-necked (14/20 joints) 250-mL round-bottomed flask is equipped with a Teflon-coated magnetic stir bar (2 cm, football-shaped) and a jacketed reflux condenser (14/20 joint). The top of the condenser and the second neck of the round-bottomed flask are capped with rubber septa after the apparatus is flame-dried under reduced pressure (<0.1 mm Hg) and cooled to 23 ℃ under a nitrogen atmosphere (Figure 1A). Both pieces of glassware are brought into the glovebox (Notes 2 and 3).
Inside the glovebox, the reaction flask is charged sequentially with bis(pinacolato)diboron (1) (6.00 g, 23.6 mmol, 1.00 equiv) (Note 4), 4,4'-di-tert-butyl-2,2'-dipyridine (DTBP) (127 mg, 473 μmol, 0.020 equiv) (Note 5) and (1,5-cyclooctadiene)(methoxy)iridium(I) dimer ([Ir(COD)OMe]2) (157 mg, 236 μmol, 0.010 equiv) (Note 6). The condenser is fitted to the reaction flask, and secured using a Keck clip. The assembled and septa-sealed glassware is transferred out of the glovebox (Note 7), and into a fume hood (Figure 1B). The septum on top of the reflux condenser is pierced with a needle (18G) that leads to a nitrogen manifold which is connected to a bubbler, allowing for a positive flow of nitrogen gas. The atmosphere is maintained under a slight positive pressure of nitrogen for the duration of the reaction. Under a positive pressure of nitrogen, the Keck clip is removed, and the joint between the flask and condenser is sealed with Teflon tape, then the Keck clip is mounted again. Cyclohexane (39 mL, 0.60 M) is added to the reaction vessel via a plastic syringe fitted with an 18G needle through the rubber septum on the second neck of the flask (Note 8). Triethylsilane (2) (15.1 mL, 94.5 mmol, 4.0 equiv) is added (Figure 1C) via a plastic syringe fitted with an 18G needle through the same septum (Notes 9 and 10). The septum that had been pierced is wrapped with electrical tape. The condenser is attached to hosing connected to a tap and a drain. The water is turned on, and the reaction flask is then placed in an oil bath set to 80 ℃ (Figure 1D) (Note 11). The flask is covered with aluminum foil (Figure 1E) and stirred at 700 rpm for 16 h.

Figure 1. A. Set-up of glassware for the reaction; B. Solid reagents in flask; C. Mixture after addition of cyclohexane and triethylsilane (2); D. Reaction mixture at 0 h; E. Reaction set-up with foil for insulation; F. Reaction mixture after 16 h at 80 ℃ (Photos provided by checkers)
After stirring for 16 h, the reaction flask is removed from the oil bath and allowed to cool to 23 ℃ (Figure 1F). An aliquot was taken from the reaction mixture and the reaction is determined to be complete by TLC analysis (Note 12). The reflux condenser is removed from the flask, and the reaction mixture is transferred to a single-necked (24/40 joint) 250-mL round-bottomed flask utilizing a funnel. The initial reaction flask was rinsed 3 times with 3 mL of diethyl ether which were transferred to the other flask. The reaction mixture was concentrated under reduced pressure via a rotary evaporator (40 ℃ bath, 350 mbar to 50 mbar). The flask is placed under high vacuum for 30 seconds to yield the crude product as a dark red oil (Note 13).
A silica gel column (4 cm OD x 16 cm tall) is prepared with 80 g silica gel (Note 14) wetted with 250 mL of hexanes (Note 15). The crude product is then directly loaded onto the column (Note 16), rinsing the flask that contained crude material 3 times with 2 mL of diethyl ether (Note 17), followed by loading the rinses onto the column. The column is then topped with a 2 cm high layer of sand (Figure 2A). The column is eluted with 500 mL of 19:1 hexanes:diethyl ether. Fraction collection (16 x 125 mm culture tubes, 15 mL fractions) begins immediately. The eluate containing the product (Note 18) is concentrated under reduced pressure via a rotary evaporator (40 ℃ bath, 350 mbar to 100 mbar), to yield the crude product as a black oil (Figure 2B).
Figure 2. (A) Column loaded with crude material; (B) Material after column purification (Photos provided by authors)
The material is transferred to an oven-dried 25-mL round-bottomed flask with a Teflon-coated dry magnetic stir bar (2 cm, football-shaped). The initial flask containing the crude material is rinsed with 3 mL of diethyl ether and the material is transferred to the 25-mL round-bottomed flask. The reaction mixture is concentrated under reduced pressure via a rotary evaporator (40 ℃ bath, 350 mbar to 50 mbar), and the flask is placed under high vacuum for 30 seconds (Note 19).
The 25-mL round-bottomed flask is equipped with an oven-dried (14/20 joint) one-piece short-path vacuum distillation apparatus, thermometer and a three-necked "cow-type" distillation receiver connected to three oven-dried 25-mL flasks, with all the joints secured by Keck clips (Figure 3A). Water circulation and stirring (550 rpm) are initiated. The apparatus is placed in an oil bath heated to 190 ℃, and put under vacuum (100 mbar) (Note 11 and 20). The distillation apparatus is insulated with 4 layers of aluminum foil (Figure 3B) (Note 21). When the thermometer reads 145 ℃, distillation proceeded at a constant rate (Note 22, 23, and 24).
Figure 3. A. Setup for distillation; B. Insulated distillation apparatus (Photos provided by the authors)
Once distillation is complete, the apparatus is taken out of the oil bath and left to cool to 23 ℃. Once at ambient temperature, the apparatus is disconnected from the vacuum, and the 25 mL flask containing the product was removed (Note 25). This provided triethyl(4,4,5,5-tetramethyl-1,3,2 dioxaborolan-2-yl)silane (3) as a clear and colorless oil (3.35 g, 13.6 mmol, 57.7% yield, 98.6% pure) (Figure 4) (Notes 26, 27, and 28).
Figure 4. Distilled triethyl(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)silane (3) (Photo provided by authors)
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/about/governance/committees/chemical-safety.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
bis(pinacolato)diboron (
1),
4,4'-di-tert-butyl-2,2'-dipyridyl (
DTBP),
(1,5-cyclooctadiene)(methoxy)iridium(I) dimer (
[Ir(COD)OMe]2),
cyclohexane, and
triethylsilane (
2).
2. After cooling to 23 ℃ under a
nitrogen atmosphere, the condenser was removed from the round-bottomed flask, and both pieces of glassware were immediately brought into the glovebox. A Keck clip was also brought into the glovebox to secure the condenser before the reaction setup is removed from the glovebox.
3. A
nitrogen-filled glovebox is used to store and load reagents due to the oxygen sensitivity of
(1,5-cyclooctadiene)(methoxy)iridium(I) dimer (
[Ir(COD)OMe]2) and
4,4'-di-tert-butyl-2,2'-dipyridyl (
DTBP).
4.
Bis(pinacolato)diboron (
1) (98.5%) was purchased from Ambeed and used as received.
5.
4,4'-Di-tert-butyl-2,2'-dipyridyl (
DTBP) (98%) was purchased from Ambeed and used as received.
6.
(1,5-Cyclooctadiene)(methoxy)iridium(I) dimer (
[Ir(COD)OMe]2) (100%) was purchased from Sigma-Aldrich and used as received.
7. The reflux condenser is attached to the reaction flask while in the glovebox to avoid air exposure.
8.
Cyclohexane (HPLC grade, 99%) was purchased from Fisher Scientific and used as received.
9.
Triethylsilane (
2) (99%) was purchased from Sigma-Aldrich and used as received.
10.
Triethylsilane (
2) must be used in excess for optimal yield. When the reaction is performed with fewer equivalents of
2, the yield is decreased.
11. The authors used a metal heating mantle instead of an oil bath.
12. The progress of the reaction is monitored by TLC analysis. After allowing the reaction flask to cool to 23 ℃, an aliquot (20 μL) was taken from the reaction mixture using a 1 mL plastic syringe fitted with a 18G long needle. The reaction mixture is then spotted on a silica gel TLC plate, and a solvent system of 9:1 hexanes:
ethyl acetate is used as an eluent. The plate is visualized using potassium permanganate solution.
Bis(pinacolato)diboron (
1) has R
f =0.4 and
triethyl(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)silane (
3) has R
f = 0.7 (Figure 3), with a byproduct at R
f = 0.6.
Figure 5. TLC of the crude reaction mixture after 16 h (S = bis(pinacolato)diboron (1), C = co-spot of S and R, R = reaction mixture) (Photo provided by authors)
13. The reaction mixture is placed under high vacuum for 30 seconds to ensure most of the excess
triethylsilane is removed before chromatography.
14. Silica gel (technical grade, pore size 60 Å, 230 - 400 mesh particle size, 0.040-0.063 nm particle size) was purchased from Sigma-Aldrich and used as received.
15. Hexanes (98.5%) was purchased from Fischer Scientific and used as received.
16. The crude product oil mixture bubbles when it is loaded onto the column, likely due to the dehydrogenative decomposition of excess silane. Caution must be taken.
17.
Diethyl ether (Anhydrous, 99%) was purchased from Sigma-Aldrich and used as received.
18. Fractions containing desired product were identified via TLC. Using a solvent system of 19:1 hexanes:
ethyl acetate, compound
3 has a R
f 0.7. Fractions 13-25 contained the desired product and were collected in a 500 mL round-bottomed flask. Fractions 13-16 were collected even though they contained an impurity with a R
f = 0.4 since the impurity can be removed via fractional distillation. Each fraction was rinsed with
diethyl ether (2 x 1 mL) and collected, to ensure quantitative transfer.
Figure 6. Fractions collected (Photo provided by authors)
19. To ensure bumping does not occur in the distillation process, solvent should not be present in the reaction mixture. The reaction mixture is placed under high vacuum for 30 seconds to ensure this.
20. To ensure a vacuum pressure of 100 mbar is maintained, the authors connected the one-piece short path vacuum distillation apparatus to a rotary evaporator. The checkers connected the distillation apparatus to a Schlenk line under high-vacuum. The pressure was finely controlled by a needle value and vacuum gauge which were also connected to the Schlenk line.
21. Four layers of aluminum foil are utilized to minimize the dissipation of heat.
22. Once the temperature at the thermometer reads >130 ℃ and a drop has distilled into one of the 25 mL flasks, the cow is rotated to collect in a new 25 mL flask. Collecting the first drop of product that distills over has been empirically found to decrease purity.
23. The checkers found that heating the apparatus with an oil bath to 190 ℃ avoided rapid product distillation and enabled more precise collection of a single drop of product. After the cow is rotated to a new 25 mL collection flask, the apparatus is heated to 220 ℃.
24. Distillation is stopped once the temperature at the thermometer falls below 100 ℃ and only a black residue remains in the distilling flask.
25. Both column purification and fractional distillation are needed to ensure high purity of the product.
26. The purity of product
3 obtained from distillation was determined to be 98.6% by qNMR
pdf using
1,3,5-trimethoxybenzene (Oakwood Chemical, 98%) as the internal standard.
27. The product was characterized as follows:
1H NMR
pdf (500 MHz, CDCl
3) δ: 1.22 (s, 12H), 0.96 (t,
J = 7.9 Hz, 9H), 0.59 (q,
J = 7.9 Hz, 6H);
13C NMR
pdf (125 MHz, CDCl
3) δ: 83.0, 25.2, 8.5, 3.1; IR (neat): 2951, 2872, 1457, 1370, 1294, 1136, 1015, 960, 849, 714, 699, 588 cm
-1; HRMS-DART (m/z) [M + H]
+ calc'd for C
12H
28BO
2Si, 243.1946; found, 243.1948; R
f = 0.7 (9:1 hexanes:
ethyl acetate).
28. Additional runs on the same scale provided yields of 55-58% with purities >98 wt% as determined by qNMR.
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
This procedure provides a simple protocol for the synthesis of trialkylsilylboranes, which are valuable reagents for the synthesis of organosilicon and organoboron compounds.
3 Currently, the methods available for accessing silylboranes remain limited.
4 Common methods to synthesize silylboranes require treating silyllithium reagents with boranes, which make accessing trialkylsilylboranes more challenging.
3,4 The iridium-catalyzed borylation of silanes developed by Hartwig and coworkers
5 is attractive as it avoids the use of silyllithium species, and provides a method to access alkyl silylboranes.
The one-step methodology by Hartwig to obtain silylboranes provides a strategy to prepare a series of trialkylsilylboranes
5 by leveraging iridium catalysis (Figure 7).
5 As shown in Figure 7, various silyl substituents are tolerated to provide silylboranes (
3, 6-9). However, the use of triisopropylsilane is not viable, as attempts to synthesize
10 were unsuccessful.
Figure 7. Scope of borylation of silanes
As shown in Figure 8, triethylsilylborane
3 can be used to achieve borylation or silaboration transformations. For example, aryl boronic ester
12 and primary benzyl boronic ester
14 are accessible via iridium-catalyzed C-H borylation of
11 and
13, respectively, utilizing triethylsilylborane
3 as the boron source.
5,6 Additionally,
3 can be implemented in the stereo- and regioselective silaboration of alkynes.
7 This is exemplified in the stereoselective base-catalyzed 1,1-silaboration of terminal alkyne
15 to obtain (Z)-silylborane
16. 1,2-Silaboration of nitrogen-containing heteroarenes is also possible, as illustrated in the silaboration of quinoline
17 to form
N-boryl-2-silyl-1,2-dihydroquinoline
18.
8Figure 8. Selected applications of Et3SiBpin (3) for borylation and silaboration
Triethylsilylborane
3 is also a robust reagent for silylation. Depending on the source of activation, diverse intermediates and products can be formed.
4 For example, in the presence of a photoredox catalyst under blue LED irradiation, trialkylsilylborane
3 can undergo a single-electron oxidation process, resulting in the formation of a silyl radical. The silyl radical can then participate in different modes of reactivity, such as the hydrosilylation of alkene
19 to access
20 (Figure 9).
9 Triethylsilylborane
3 can also participate in transition metal-catalyzed silylation reactions.
4 This is seen in the nickel/copper dual-catalyzed decarboxylative silylation of ester
21, giving rise to silylated benzofuran
22.
10 Notably, the reagent can also be harnessed to form products with stereocenters. This is demonstrated in the copper-catalyzed diastereo- and enantioselective addition of the silyl group to cyclopropene
23 to yield silyl-substituted cyclopropane
24 in 99% enantiomeric excess.
11 Another example involves the regioselective opening of epoxides. In this case, a lithiated silane species can be generated in situ from Et
3SiBpin by the action of MeLi. Subsequent nucleophilic addition to epoxide
25 gives silyl alcohol
26 en route to a strained heterocyclic allene precursor.
12
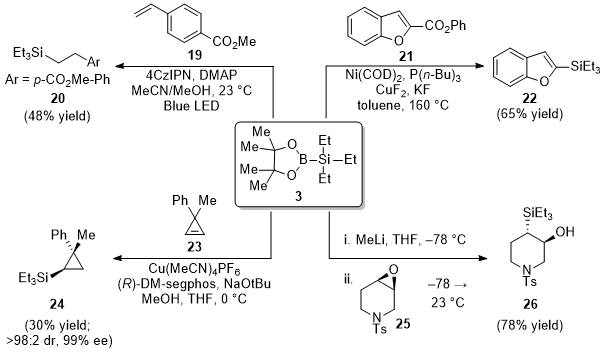
Figure 9. Selected applications of Et3SiBpin (3) for silylation of diverse substrates
Overall, this procedure provides a means to access trialkylsilylboranes via the iridium-catalyzed borylation of silanes. This one-step strategy developed by Hartwig is attractive as it utilizes readily available starting materials and a low catalyst loading. The development of this method has prompted further research into accessing new classes of trialkylsilylboranes.
13 This methodology, as well as related advances, will encourage the further study of the applications of silylboranes as versatile reagents to access a wide array of functionalized compounds.
Appendix
Chemical Abstracts Nomenclature (Registry Number)
bis(pinacolato)diboron; (1) (73183-34-4)
4,4'-di-tert-butyl-2,2'-dipyridyl (DTBP); (72914-19-3)
(1,5-cyclooctadiene)(methoxy)iridium(I) dimer ([Ir(COD)OMe]2); (12148-71-9)
cyclohexane anhydrous; (110-82-7)
triethylsilane; (2) (617-86-7)

|
Christina Rivera was born and raised in San Diego, California. In 2022, she received her B.S. in Chemistry emphasize in Biochemistry from San Diego State University, where she carried out research under the direction of Professor Byron Purse. In 2023, she began her graduate studies at the University of California, Los Angeles, where she is currently a second-year graduate student in Professor Neil K. Garg's laboratory. Her studies primarily focus on developing synthetic methods involved strained cyclic intermediates. |

|
Benjamin Janda was born and raised in San Jose, California. In 2023, he received his B.S. in Chemistry from Chapman University where he researched in the laboratory of Professor Allegra Liberman-Martin. In 2023, he began his graduate studies at the University of California, Los Angeles, where he is currently a second-year graduate student in Professor Neil K. Garg's laboratory. His studies focus on synthetic methods using strained cyclic allenes. |

|
Neil Garg is the Distinguished Kenneth N. Trueblood Professor of Chemistry at the University of California, Los Angeles. His laboratory develops novel synthetic strategies and methodologies to enable the total synthesis of complex bioactive molecules. |

|
James Pearson received his B.Sc. and M.Sc. in Chemistry from King's University and the University of Alberta, where he conducted research with Professors Kris Ooms and Steve Bergens. He began his PhD with Professor Sophie Rousseaux at the University of Toronto in Summer 2021, where he is currently working on photochemical rearrangements and Ni-catalyzed cross-coupling reactions. |

|
Sophie Rousseaux is an Associate Professor of Chemistry at the University of Toronto, where she also holds a Canada Research Chair (Tier 2) in Organic Chemistry and serves on the Scientific Leadership Team of the Acceleration Consortium. Her group's research interests include organic synthesis, catalysis, and organometallic chemistry, with a particular focus on the synthesis of small rings and nitrile-containing molecules. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved