1. Procedure (Note 1)
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/about/governance/committees/chemical-safety.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
2-hydroxyphenyl boronic acid,
potassium carbonate,
ethanol,
toluene,
Pd(PPh3)4,
pentafluorobromobenzene,
HCl (1M),
dichloromethane, anhydrous
sodium sulfate,
ethyl acetate, silica gel,
cyclohexane,
para-formaldehyde,
magnesium chloride,
tetrahydrofuran,
triethylamine,
Candida antarctica lipase B (CAL-B) immobilized on polystyrene,
cis-1,2-diaminocyclohexane,
diallyl carbonate,
methanol,
ammonia,
nickel(II)acetate tetrahydrate,
sodium borohydride, and anhydrous
magnesium sulfate, as well as hazards associated with nickel hydride reductions,
i.e. sodium borohydride reductions carried out in the presence of catalytic amounts of nickel salts.
2. The checkers used both absolute
ethanol stored under inert atmosphere (anhydrous) and absolute
ethanol (HPLC grade) stored under air without distillation. Both gave the product mixture in the same yields (74%).
3. The authors obtained absolute
toluene (99.85%) and THF (99.5%, stabilized) from Arcos Organics (extra dry over molecular sieves, AcroSeal grade). Absolute
ethanol (99.8%) was purchased from Fisher Scientific. All other solvents were obtained in technical grade and were distilled (no inert gas and no drying agents applied) prior to use. The checkers purchased absolute
toluene (anhydrous, 99.8%) from Alfa Aesar and
ethanol (anhydrous, ≥99.5%) from Sigma Aldrich and both were used as purchased; absolute THF was purchased from VWR and then degassed and stored under argon and molecular sieves prior to use;
methanol (ACS grade) and
DCM (ACS grade) used in step D were purchased from VWR and Fisher Chemical, respectively, and used as received. Solvents for work-ups and purifications were obtained in ACS grade and used as received.
4. The authors purchased reagents used in step A from the following companies: boronic acid
1 (98%) and
pentafluorobromobenzene (
2, 98%): BLDPharm;
K2CO3 (anhydrous, 99%): Fisher Chemical;
Pd(PPh3)4: Carbolution (no purity given, product number: CCC03013). Checkers purchased boronic acid
1 (98%) from Ambeed, pentafluorobromobenzene (
2, >99%) from TCI,
K2CO3 (anhydrous, 99%) from Alfa Aesar, and
Pd(PPh3)4 (99%) from Strem.
5. For TLC analysis, VWR TLC aluminium-backed plates Silica F254 (200 μm layer thickness) 20x20 cm were used and cut to appropriate size. For the elution of the plates, standard TLC chambers were used. Eluent compositions and detection methods are stated in the
Notes for all individual experimental procedures.
6. TLC of the reaction mixture [
c-hex/
EtOAc 9:1, UV light (λ = 254 nm)/
KMnO4 dye: 3 g
KMnO4, 20 g
K2CO3, 5 mL 5%
NaOH solution and 300 mL water].
Figure 7. TLC of the reaction mixture. B: boronic acid 1, R: reaction mixture in two different concentrations, F: pentafluorobromobenzene (2) (Photos provided by the authors)
7. Silica gel for column chromatography (0.035-0.07 mm) was purchased from Acros.
8. Column size: 8x11 cm, 270 g of silica gel, elution with ca. 500 mL of solvent, then collection of fractions (50 mL each) was started. Checkers collected fractions 7-30.
9. The phenol by-product can be removed by recrystallisation from
c-hex. However, it is more convenient to continue on with the mixture, and to perform the purification after the formylation step. The phenol impurity is readily detected in the mixture by its characteristic
1H-NMR signals: (300 MHz, CDCl
3) δ (ppm) = 7.29 - 7.23 (m, 2H), 6.99 - 6.91 (m, 1H), 6.90 - 6.82 (m, 2H), 5.52 (bs, 1H, OH). See below for the analytical data of the pure, recrystallized
2',3',4',5',6'-pentafluoro-2-hydroxy-(1,1'-biphenyl) (
3). For the recrystallization, the 5:1 mixture of
3 with phenol was dissolved, at reflux temperature, in the minimal amount of
c-hex necessary for full dissolution. After cooling to room temperature and after completion of the crystallization, the colorless crystals of
3 were isolated by suction filtration using a 60-mL medium-porosity Büchner funnel and dried
in vacuo (ca. 1 mbar) at room temperature (18-22 ℃). Checkers obtained white crystals which became a powder
in vacuo; percent of recovery was 41%.
Analytical data of recrystallized 3: mp. 100 ℃; 1H NMR pdf (600 MHz, CDCl3) δ 7.36 (t, J = 7.8 Hz, 1H), 7.23 (d, J = 7.6 Hz, 1H), 7.06 (t, J = 7.0 Hz, 1H), 6.94 (d, J = 8.1 Hz, 1H), 5.06 (s, 1H); 13C NMR pdf (151 MHz, CDCl3) δ 153.4, 144.6 (d, J = 248 Hz, 2C), 140.9 (d, J = 254 Hz), 137.8 (d, J = 252 Hz, 2C), 132.1, 131.3, 121.3, 116.3, 113.7, 112.2 (d, J = 4 Hz); 19F NMR pdf (565 MHz, CDCl3) δ -139.83 (dd, J = 23.0, 8.1 Hz), -155.21 (t, J = 20.8 Hz), -162.45 (td, J = 22.1, 8.2 Hz); HRMS [M•]+ calculated for C12H5F5O: 260.0261, found: 260.0271; IR pdf (ATR): 3513, 1485, 1444, 1059, 982, 870, 757 cm-1; The analytical data are in agreement with the literature.3 The purity of recrystallized 3 was determined to be 98.7 wt% as determined by qNMR pdf using methyl 3,5-dinitrobenzoate (99%, Sigma-Aldrich) as an internal standard.
Figure 8. Pure compound 3 after recrystallization. (Photo provided by the checkers)
10. The authors obtained reagents used in step B from the following companies:
para-formaldehyde (≥95%) and
MgCl2 (≥98%, anhydrous, reagent grade): Sigma Aldrich;
Et3N (≥99%, laboratory reagent grade): Fisher Scientific. Checkers purchased
para-formaldehyde (≥90.0%) from TCI,
MgCl2 (97.5%, 2% max
H2O) from Strem, and
Et3N (anhydrous, ≥99.5%) from Sigma Aldrich.
11. Crude TLC of the reaction was performed using two concentrations of unpurified reaction mixture [
c-hex/
EtOAc 9:1, UV light (λ = 254 nm)].
Figure 9. TLC of the reaction mixture of step B. S: starting material 3, C: crude mixture (Photos provided by the checkers)
12. Checkers transferred the reaction mixture to a one-neck flask before concentration with a rotary evaporator and continued with the same flask for
HCl dropwise addition.
13. Column size: 13x5.5 cm, 170 g of silica gel; 600 mL of a 2:1 mixture of
c-hex/
toluene, followed by 500 mL of a 1:1 mixture was used as eluent. Ca. 250 mL of solvent were eluted, then collection of fractions (50 mL each) was started. Product: fractions 4 to 18. Checkers used ca. 200 g silica gel with 650 mL of 2:1 mixture of hexanes/
toluene then 1:1 mixture; checkers collected fractions 9 to 21.
Figure 10. A. Column chromatography; B. TLCs of the collected fractions [1:1 hexanes/toluene]. (Photos provided by the checkers)
14. Analytical Data of
4: mp. 117 ℃;
1H NMR
pdf (500 MHz, CDCl
3) δ 11.45 (s, 1H), 9.98 (d,
J = 1.0 Hz, 1H), 7.73 (dd,
J = 7.7, 1.7 Hz, 1H), 7.52 (d,
J = 7.6 Hz, 1H), 7.17 (t,
J = 7.6 Hz, 1H);
13C NMR
pdf (126 MHz, CDCl3) δ 196.5, 159.5, 144.6 (d,
J = 252 Hz, 2C), 141.2 (d,
J = 254 Hz, 2C), 139.0, 137.8 (d,
J = 251.2 Hz), 135.5, 121.1, 120.0, 115.7, 110.7;
19F NMR
pdf (565 MHz, CDCl3) δ -139.56 (dd,
J = 23.0, 8.3 Hz), -154.30 (td,
J = 21.2, 8.5 Hz), -162.22 (td,
J = 21.5, 8.1 Hz); HRMS [M•]
+ calculated for C
13H
5F
5O
2: 288.0210, found: 288.0210; IR
pdf (ATR): 3031, 2363, 1647, 1490, 989, 962, 739, 666 cm
-1; The analytical data are in agreement with the literature.
3 The checkers determined the purity of recrystallized
4 to be 100 wt% as determined by qNMR
pdf using methyl 3,5-dinitrobenzoate (99%, Sigma-Aldrich) as an internal standard. The authors confirmed the purity by elemental analysis: Anal. Calcd for C
13H
5F
5O
2: C 54.18, H 1.78; Found: C 54.57, H 1.85.

Figure 11. A. Formation of needle shaped crystals from cyclohexane in a 100 mL RBF; B. Pure compound 4 after recrystallization. (Photos provided by the checkers)
15. The reagents used by both authors and checkers in step C were purchased from the following companies:
cis-DACH (98%): BLDPharm;
diallyl carbonate (99%) and CAL-B (product number 52583,
Candida antarctica lipase B, recombinant from yeast, immobilized on Immobead 150, ≥ 2000 U/g): Sigma Aldrich.
16. The authors did not dry or evacuate the flask prior to flushing with argon. However, because anhydrous
toluene and inert atmosphere were employed by the authors, checkers decided to flame dry the flask and exchange atmosphere with argon.
17. After reaction setup, checkers closed the outlet of the Schlenk flask and stirred the reaction under inert atmosphere without active flow of argon. The authors stirred the reaction under a constant flow of argon. Checkers noticed the occurrence of a suspended white material on day 2 of stirring..
18. The original procedure from the authors was a 4-day reaction. But checkers observed much slower reactivity. After communication with the authors, the procedure was modified.
19. TLC was performed using an aliquot of the reaction mixture (obtained under positive pressure) after stirring for 7 days.
KMnO4 was used for detection [95:5
DCM:7M NH
3 in
MeOH] (see Note
6 and
20).
Figure 12. TLC of the reaction mixture after staining with KMnO4. SM: starting material 5, C: crude mixture (Photo provided by the checkers)
20. Column size: 16x5 cm, 200 g of silica gel. Ca. 150 mL of solvent were passed through the column, then collection of fractions (50 mL each) was started. Product: fractions 16 to 32.
KMnO4 was used for detection (see Note
6). The authors saturated
MeOH through bubbling NH
3 for 3 h at 0 ℃. Checkers purchased 7M NH
3 in
MeOH from Oakwood Chemicals. For collection of the product, checkers collected fractions 10 to 24.
Figure 13. A. TLC of the reaction mixture of step C after stirring for 10 d; B. Column chromatography; C. TLCs of the collected fractions [95:5 DCM:7M NH3 in MeOH]. (Photos provided by the checkers)
21. Analytical data of
6: [α]
D20 4.3 (c 1.7, CHCl
3);
1H NMR
pdf (500 MHz, CDCl
3) δ 5.99 - 5.83 (m, 1H), 5.34 - 5.22 (m, 2H), 5.20 (dq,
J = 10.5, 1.4 Hz, 1H), 4.54 (d,
J = 5.7 Hz, 2H), 3.74 - 3.54 (m, 1H), 3.01 (dt,
J = 6.9, 3.8 Hz, 1H), 1.71 - 1.57 (m, 2H), 1.56 - 1.26 (m, 8H);
13C NMR
pdf (126 MHz, CDCl3) δ 156.1, 133.2, 117.7, 65.5, 52.2, 49.9, 32.3, 28.1, 23.1, 20.8; HRMS [M+Na]
+ calculated for C
10H
18N
2O
2Na: 221.1266, found: 221.1265; IR
pdf (ATR): 3326, 2929, 2858, 2362, 1698, 1522, 1236, 1040 cm
-1; The analytical data are in agreement with the literature.
4 Please note the authors reported the clean product (based on both NMR analysis and elemental analysis; Anal. Calcd for C
10H
18N
2O
2: C 60.58, H 9.15, N 14.13; Found: C 60.50, H 9.05, N 14.16) as a pale yellow oil. However, the checkers obtained
6 as a dark brown oil with a purity of only 89.2 wt% by qNMR
pdf using methyl 3,5-dinitrobenzoate (99%, Sigma-Aldrich) as an internal standard. The peak for the unidentifiable impurity was at δ 3.62 ppm in the
1H NMR (overlapped with
6) and δ 70.8 ppm in the
13C NMR
pdf.
22. The checkers determined the enantiomeric excess to be >99 % by HPLC analysis (Figure 14). For this purpose, the product
6 was derivatized with 1-isothiocyanato-3,5-bis(trifluoromethyl)benzene: 0.20 g (1.0 mmol, 1.0 equiv) of sample was dissolved in 2.2 mL THF in a flame-dried 10 mL round-bottomed flask under Ar. The isothiocyanate (0.18 mL, 1.0 mmol, 1.0 equiv) was added and the reaction was stirred overnight at room temperature. After removing solvent, the derivatized product (
S1) was purified through column chromatography (1%
MeOH/
DCM) as a yellow solid (96%). 2 mg of
S1 was dissolved in
i-PrOH and submitted to HPLC analysis. The column temperature was set to 19
oC. Diode array detection (DAD) was used, and λ = 230 nm was chosen for peak integration. HPLC:
𝜏R [min] = 3.8 (minor; 1
S,2
R), 4.7 (major; 1
R,2
S) (Diacel Chiralpak AD-H (5 mm, 4.6x100 mm),
n-hex/
i-PrOH, 95:5; flow rate = 0.8 ml/min). For preparation of racemic
S1,
5 (0.20 g, 1.8 mmol, 1.0 equiv) was dissolved in
DCM (8.8 mL) followed by addition of
Et3N (0.29 mL, 2.1 mmol, 1.2 equiv) and Alloc-Cl (0.19 mL, 1.8 mmol, 1.0 equiv). The reaction was stirred overnight at room temperature and then the solvent was removed. The racemic
S1 was purified through the same conditions (see
Note 20) and then further derivatized as stated above.
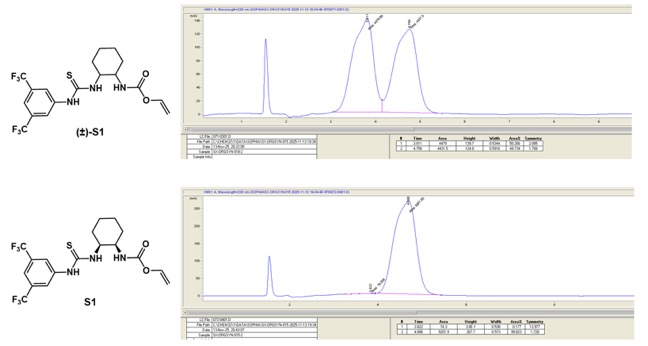
Figure 14. HPLC Comparison of racemic and enantiomerically pure 6 after derivatization.
The authors determined the enantiomeric excess of 6 to be 97% by capillary GC on chiral stationary phase. For this purpose, the product was derivatized with trifluoroacetic anhydride: 5 mg of sample were dissolved in 1.0 mL DCM in a 2-mL GC-vial. 0.5 mL sat. K2CO3 solution and a 3 mm rod-shaped Teflon-coated magnetic stir bar were added. While stirring, 3 drops of trifluoroacetic anhydride were added. The solution was stirred at room temperature for 30 min. The organic phase was then separated with a pipette, and filtered through anhydrous Na2SO4 (in a pipette with a cotton plug and a layer of Na2SO4 on top) into another GC-vial. The vial was filled up with DCM and submitted to GC analysis. 𝜏R = 31.5 min (major; 1R,2S), 33.9 (minor, 1S,2R); CP-Chirasil-Dex CB 0.25 μm, 25 m × 0.25 mm ID; 19.78 psi N2 gas; 60 ℃, 10 ℃/min to 150 ℃ (30 min).
23. This procedure can just as well be performed in an equally sized beaker (250 mL).
24. The authors reported a colorless-to-yellow color change when adding
6. Since the checkers used the dark brown oil of
6, the solution was yellow to start with and no color change was observed when adding aldehyde
4.
25. The authors reported completion of the consumption of aldehyde
4 by TLC (
c-hex/
ethyl acetate, 3:1; R
f (aldehyde
4) = 0.59, detection with
KMnO4 solution and UV light (λ = 254 nm) (see
Note 6). Because checkers were not able to obtain highly pure
6, aldehyde
4 was not fully consumed, even with extended reaction time (4.5 h) so stirring for this step was only kept at 3 h.
Figure 15. TLC of the reaction mixture before addition of Ni(OAc)2 [c-hex/ethyl acetate, 3:1]. C: crude mixture; SM: starting material 4 (Photo provided by the checkers)
26. The authors obtained the reagents used in step D from the following company: nickel(II) acetate tetrahydrate (≥99%) and
NaBH4 (≥98%): Sigma Aldrich. Checkers purchased nickel(II) acetate tetrahydrate (99%) from thermo scientific and NaBH
4 (≥98%) from Sigma Aldrich.
27. The evolution of hydrogen makes the excess of
NaBH4 necessary.
28. If a greenish solid precipitated after the color change to yellowish-green, it was dissolved by addition of more
MeOH (up to ca. 10 mL).
29. The authors used a pipet to add the water.
30. A dark orange oil. TLC analysis of the crude product showed two spots [
c-hex/
ethyl acetate, 4:1; detection with
KMnO4 solution and UV light (λ = 254 nm) (see
note 5)]: R
f = 0.38 (ligand
7) and R
f = 0.20 [inseparable mixture of the non-deprotected intermediate
8 and its reduction product (R =
n-propyl)]). Checkers' crude TLC showed four to five spots [hexanes/
ethyl acetate, 4:1; detection with UV light (λ = 254 nm)] with the largest one being the ligand.
Figure 16. TLC of the reaction mixture of step D. (Photo provided by the checkers)
31. Column size: 12x5.5 cm, 160 g of silica gel, elution with ca. 150 mL of solvent, collection of fractions (50 mL each) was started when the eluent turned yellow. Product: fractions 2 to 10 (colored yellow). Checkers collected fractions 3 to 12. The product from the column became a yellow foam
in vacuo.
Figure 17. A. Column chromatography; B. TLC of the collected fractions; C. Appearance of the product from the column in vacuo. (Photos provided by the checkers)
32. The final recrystallization from
c-hex is necessary to obtain the ligand
7 in analytically pure form and with ≥ 99 % ee. The ligand
7 displays the following physical and spectroscopic properties: bright yellow crystals; mp. 167 ℃ (163
oC reported by the authors); [α]
D20 -94.3 (c 1.2, CHCl
3); the authors reported [α]
D20 -181.9 (c 1.8, CH
2Cl
2);
1H NMR
pdf (500 MHz, CDCl
3) δ 14.01 (s, 1H), 8.53 (s, 1H), 7.44 (dd,
J = 7.7, 1.7 Hz, 1H), 7.31 (dd,
J = 7.2, 1.2 Hz, 1H), 7.14 (d,
J = 8.3 Hz, 1H), 7.09 (dd,
J = 7.5, 1.6 Hz, 1H), 7.02 (t,
J = 7.6 Hz, 1H), 6.88 (t,
J = 7.6 Hz, 1H), 4.14 (d,
J = 14.2 Hz, 1H), 4.03 (d,
J = 14.2 Hz, 1H), 3.72 (q,
J = 3.4 Hz, 1H), 2.81 (dt,
J = 11.4, 3.6 Hz, 1H), 2.01 - 1.33 (m, 8H);
13C NMR
pdf (126 MHz, CDCl
3) δ 165.3, 159.5, 156.6, 145.6, 143.6 (2C), 141.6, 139.9, 138.8 (2C), 136.8 (2C), 134.8, 133.4, 130.9, 130.0, 122.9, 119.2, 119.0, 118.7, 115.0, 114.2, 112.9, 112.0, 68.1, 57.2, 49.4, 33.1, 27.6, 24.9, 20.7;
19F NMR
pdf (471 MHz, CDCl
3) δ -139.34 - -139.47 (m), -139.47 - -139.59 (m), -139.88 (dd,
J = 23.5, 8.1 Hz), -140.05 (dd,
J = 23.5, 8.2 Hz), -155.50 (t,
J = 20.9 Hz), -156.46 (t,
J = 20.9 Hz), -162.70 (td,
J = 23.2, 8.3 Hz), -162.88 - -163.46 (m); HRMS [M+Na]
+ calculated for C
32H
22F
10N
2O
2Na: 679.1419, found: 679.1443; IR
pdf (ATR): 2939, 2864, 2358, 1628, 1520, 1495, 1447, 981 cm
-1.

Figure 18. A. Formation of crystals of 7 from cyclohexane (after 1 week); B. Appearance of the final recrystallized ligand 7. (photos provided by the checkers)
33. The checkers determined the purity of recrystallized
7 to be 100 wt% as determined by qNMR
pdf using methyl 3,5-dinitrobenzoate (99%, Sigma-Aldrich) as an internal standard. The authors confirmed the purity by elemental analysis (Anal. Calcd for C
32H
22F
10N
2O
2: C, 58.54; H, 3.38; N, 4.27. Found: C, 58.74; H, 3.47; N, 4.09.). HPLC:
𝜏R [min] = 9.0 (major,
7), 13.1 (minor,
ent-
7) (Diacel Chiralpak OD-H (5 μm, 4.6x100 mm),
n-hex/
i-PrOH, 95:5; flow rate = 0.5 ml/min). For the determination of enantiomeric purity, ca. 1 mg of the solid ligand
7 was placed in a 2 mL vial and was dissolved in ca. 0.4 mL
i-PrOH. If necessary, ultra-sonication at ca. 20
oC can be applied. Upon complete dissolution of the solid, 1.5 mL
n-hex was added and the solution was submitted to HPLC-analysis (auto-injection). The column temperature was set to 19
oC. Diode array detection (DAD) was used, and λ = 254 nm was chosen for peak integration. For HPLC assay, checkers received a racemic sample of
7 from the authors. The authors reported the preparation was done following step D with racemic
6 (Figure 19).
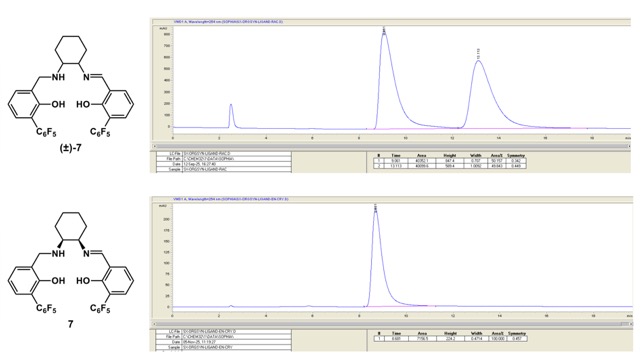
Figure 19. HPLC Comparison of racemic and enantiomerically pure 7
3. Discussion
Scheme 2. (a) Self-assembly of the dimeric Ti-epoxidation catalyst 11 from the cis-DACH salalen ligand 7 and Ti(OiPr)4; (b) asymmetric epoxidation of 1-decene (12) using the in situ generated catalyst 11
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved