Org. Synth. 2026, 103, 78-100
DOI: 10.15227/orgsyn.103.0078
Synthesis of N-Pyridinium Aziridines
Submitting Authors: Samya Samanta, Hao Tan, Saikat Chatterjee, David C. Powers*
1Checking Authors: Michael M. Gilbert and Nathan D. Ide*
21. Procedure (Note 1)
A. 2,4,6-Triphenyl-[1,2'-bipyridin]-1-ium tetrafluoroborate (1). A 500-mL single-necked (24/40) round-bottom flask equipped with a round Teflon-coated stir bar (37 mm × 9.5 mm) is charged with 2,4,6-triphenylpyrylium tetrafluoroborate (8.00 g, 95%, 20.2 mmol, 1.0 equiv) (Note 2), using a polypropylene anti-static weighing funnel, and dichloromethane (200 mL) (Note 3), in two portions via 100 mL glass graduated cylinder, to obtain a bright yellow suspension (Figure 1A). 2-Aminopyridine (2.85 g, 99%, 30.3 mmol, 1.5 equiv) (Note 4) is added, using a polypropylene anti-static weighing funnel, to the yellow suspension, the round-bottom flask is capped, and the reaction mixture is stirred (Note 5) at 220 RPM at room temperature (Note 6) for 16 h. During this time, the reaction mixture assumes a bright red-brown color (Figure 1B).

Figure 1. A. Reaction mixture following suspension of 2,4,6-triphenylpyrylium tetrafluoroborate in dichloromethane; B. Reaction mixture following addition of 2-aminopyridine and stirring for 16h
At this time, the magnetic stirring is stopped, and the reaction mixture is allowed to settle. The stir bar is removed from the reaction mixture. The reaction mixture is concentrated to dryness under reduced pressure (starting at 250 Torr and reducing to 25 Torr) on a rotary evaporator with the heating bath set at 25 °C (Figure 2A). The crude product is redissolved in dichloromethane (20 mL). Diethyl ether (10 mL) (Note 7) is added slowly as the antisolvent for recrystallization (Figure 2B). The round-bottom flask is capped and stored at -20 °C for 12 h.
Figure 2. A. Residue obtained after evaporating volatiles in Step A; B. Product mixture following dissolution of residue in dichloromethane and diethyl ether for recrystallization; C. Crystallization after overnight storage at -20 °C
After cooling, bright orange crystals are obtained (Figure 2C) (Note 8). The orange crystals are isolated by vacuum filtration through a 60 mL disposable 10-micron polyethylene fritted funnel (Figure 3A) (Note 9) and washed with three 20 mL portions of diethyl ether. The rinses were combined with the initial liquors. Additional precipitate was observed in the filtrate and was isolated by a second filtration. The combined solids were washed with three 20 mL portions of diethyl ether to obtain light orange crystalline solids (Figure 3B). The crystals are crushed to fine powder using a mortar and pestle (Note 10) and dried under vacuum (77 Torr, Note 11) at 40 °C for 22 h. At this point, 2,4,6-triphenyl-[1,2'-bipyridin]-1-ium tetrafluoroborate (1) is obtained (8.84 g, 93%, Note 12) (Figure 3C) and 97% purity as determined by qNMR (Note 13). The melting point of 1 is determined to be 221-224 °C.

Figure 3. A. Filtration set up for isolation of crystals. B. Light orange crystals of compound 1. C. Compound 1 as fine powder
B. 1-Amino-2,4,6-triphenylpyridin-1-ium tetrafluoroborate (2). A 250-mL single-necked (24/40) round-bottom flask equipped with a round Teflon-coated stir bar (37 mm × 9.5 mm) is charged with 2,4,6-triphenyl-[1,2'-bipyridin]-1-ium tetrafluoroborate (1, 8.50 g, 18.0 mmol, 1.0 equiv), using a polypropylene anti-static weighing funnel, (Figure 4A) and ethanol (60 mL), using a 100 mL glass graduated cylinder (Note 14). To this suspension (Figure 4B), hydrazine monohydrate (4.40 mL, >98%, 90.0 mmol, 5.0 equiv, Note 15) is added in one portion, using a 5 mL HDPE syringe with a 1.5" 21-gauge needle. A condenser (24/40) (Note 16) is attached to the reaction flask, and the reaction mixture is heated to reflux (80 °C) for 4 h (Figure 4C) with stirring (250 RPM). After 4 h, a yellow solution is observed (Figure 4D). The flask was removed from the heat source and the contents allowed to cool to room temperature (Note 6) and the stir bar is removed. Diethyl ether (3 mL) is added to the reaction flask dropwise, the flask is capped and stored at -20 °C for at least 12 h for recrystallization.

Figure 4. A. Compound 1 in a 250-mL round bottom flask; B. Reaction mixture after the addition of ethanol to 1; C. Refluxing reaction mixture; D. Yellow solution obtained after refluxing the reaction mixture
After storage at -20 °C, light orange crystals are obtained (Note 17). Crystalline solids are isolated by vacuum filtration through a disposable 60-mL, 10-micron polyethylene fritted funnel (Note 9) and washed with three 40 mL portions of diethyl ether to obtain a light orange crystalline powder (Figure 5A). This solid is transferred into a 250-mL round-bottom flask (Note 18) and is dissolved in dichloromethane (15 mL). Diethyl ether (3 mL) is added dropwise as antisolvent (Figure 5B). The flask is capped with a plastic stopper and stored at -20 °C for at least 12 h, at which time pink crystals are formed. The crystalline solids are collected by vacuum filtration through a 60-mL disposable 10-micron polyethylene fritted funnel (Note 9) and washed with 50 mL diethyl ether. Additional precipitate was observed in the filtrate and was isolated by a second filtration. The combined solids were washed with 50 mL portions of diethyl ether to obtain orange crystalline solids (Figure 5C) (Note 19). The crystals are crushed to fine powder (Note 10) and dried in a vacuum oven under vacuum (77 Torr, Note 11) at 80 °C with an N2 bleed for 24 h to remove volatiles. At this point, 1-amino-2,4,6-triphenylpyridin-1-ium tetrafluoroborate (2), which is obtained as an off-white crystalline solid (Figure 5D) (5.11 g, 69%). A second run provided 5.26 g (71%). The compound was found to be 98% purity by quantitative NMR (Note 20). The melting point of 2 is determined to be 181-185 °C.

Figure 5. A. Light orange crystals of compound 2 after the first recrystallization (EtOH/diethyl ether crystallization); B. Second recrystallization of compound 2 (dichloromethane/diethyl ether crystallization); C. Bright orange crystals after second recrystallization of compound 2; D. Off-white fine powder of compound 2
C. 2,4,6-Triphenyl-1-(2-phenylaziridin-1-yl)pyridin-1-ium tetrafluoroborate (3). A 200-mL single-necked (24/40) Schlenk flask equipped with a round Teflon-coated stir bar (37 mm × 9.5 mm) was dried in a vacuum oven at 160 °C for at least 24 h, then removed from the oven and allowed to cool to ambient temperature under vacuum. The flask is charged with 1-amino-2,4,6-triphenylpyridin-1-ium tetrafluoroborate (2, 5.00 g, 12.2 mmol, 1.00 equiv), tetrabutylammonium iodide (225 mg, 98%, 0.61 mmol, 5.00 mol%) (Note 21), and 4 Å molecular sieves (20 g) (Note 22) (Figure 6A). All solids were charged using a polypropylene anti-static weighing funnel. The Schlenk flask is closed with a rubber septum and the side arm connected to a Schlenk line (Note 23) before anhydrous CH3CN (60 mL) (Note 24) is added through the rubber septum, in two portions using a 50 mL HDPE syringe with a 5" 16-gauge needle and stirring commenced (250 RPM). Styrene (1.40 mL, 99%, 12.2 mmol, 1.00 equiv) (Note 25) is added to the reaction mixture through the rubber septum with a 1 mL HDPE syringe affixed with a 5" 21-gauge needle, in two equal portions under positive flow of N2. The reaction mixture is allowed to stir for 10 min at room temperature (Figure 6B). Iodosylbenzene (2.70 g, 12.2 mmol, 1.00 equiv) (Figure 6C, Note 26) is weighed and divided into three 1-dram glass vials in equal portions (0.90 g each) (Figure 6D). iodosylbenzene is added in three portions (each additional portion was added 5 min after the previous) under positive flow of N2. After addition of iodosylbenzene, the reaction mixture assumes a bright orange color (Figure 6E). The flask is closed with a rubber septum and stirred at room temperature for 16h.

Figure 6. A. Solid reagents and oven-dried Schlenk flask for the synthesis of N-pyridinium aziridines; B. Reaction mixture after the addition of acetonitrile and styrene; C. Iodosylbenzene before adding to reaction mixture; D. Iodosylbenzene is added in three equal portions in 5 min intervals; E. Bright orange color of the reaction mixture after adding iodosylbenzene
After 16 h, a light orange suspension is observed (Figure 7A) and the stirring is stopped. The reaction mixture is opened to air and filtered through a 120 mL disposable fritted funnel pre-packed with celite (Note 27). The solids are washed with acetonitrile (60 mL) and the rinse collected with the initial filtrate. The cake was further rinsed with 15 mL portions of acetonitrile until the rinse filtrate was clear (2 x 15 mL), The combined reddish-brown filtrate (Figure 7B) is concentrated to dryness under reduced pressure on a rotatory evaporator (starting at 100 Torr and reducing to 25 Torr) with the heating bath set to 40 °C to afford a tacky red/brown residue. The residue is redissolved in acetonitrile (20 mL). Diethyl ether (20 mL) is added as antisolvent. The round-bottom flask is capped and stored at -20 °C for at least 12 h. After the initial cooling period, an additional 30 mL of diethyl ether is added in a single portion, the flask capped, the contents swirled by hand to ensure adequate mixing of the solvent, and the flask is stored at -20 °C for at least 12 h.

Figure 7. A. Reaction mixture after 16 h of stirring in step C; B. Filtrate following Celite filtration; C. Crystal formation in the round bottom flask after recrystallization of compound 3; D. Bright yellow crystals of compound 3 after isolation; E. Light yellow fine powder of compound 3
At this time, yellow crystals are observed (Figure 7C) and are collected by suction filtration through a 10-micron disposable 110 mL polyethylene fritted funnel. The resulting crystalline solids are washed with diethyl ether (30 mL) (Figure 7D). The bright yellow crystals are crushed into fine powder using a mortar and pestle (Note 10). The powder was transferred back to the frit and washed with another 30 mL diethyl ether and dried under vacuum (77 Torr, Note 11) at 45 °C for 12 h to remove volatiles. At this point, 2,4,6-triphenyl-1-(2-phenylaziridin-1-yl)pyridin-1-ium tetrafluoroborate (3), which is obtained as a light-yellow crystalline powder (Figure 7E) (4.04 g, 65%, Note 28) Compound 3 was found to have 96% purity by quantitative NMR (Note 29). The melting point of 3 is determined to be 188-190 °C.
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out regarding each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/about/governance/committees/chemical-safety.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
2,4,6-triphenyl-[1,2'-bipyridin]-1-ium tetrafluoroborate,
pyridin-2-amine,
dichloromethane,
diethyl ether,
ethanol,
hydrazine monohydrate,
tetrabutylammonium iodide,
styrene,
acetonitrile, and
iodosylbenzene.
2.
2,4,6-Triphenylpyrylium tetrafluoroborate (95%) was purchased from Ambeed and used as received.
3. Authors:
dichloromethane (containing 40-150 ppm amylene as stabilizer, ACS reagent, ≥99.5%) was purchased from Sigma Aldrich and used as received. Checker:
dichloromethane purchased from Sigma Aldrich, ≥99.8%, suitable for HPLC, contains amylene as a stabilizer and was used as received.
4. Authors:
2-Aminopyridine (99%) was purchased from Thermo Scientific Chemicals and used as received. Checkers:
2-Aminopyridine (>99%) was purchased from Sigma Aldrich and used as received.
5. Authors: stirring throughout these procedures is kept at a constant 800 RPM using a Heidolph Hei-PLATE Mix 'n' Heat Core+ hotplate stirrer. Checker: reactions are performed using an IKA RTC basic hotplate stirrer.
6. The term "room temperature" used throughout this manuscript refers to the range of temperatures between 22 °C and 25 °C.
7.
Diethyl ether (99.0%, anhydrous, ACS reagent, contains BHT as inhibitor) was purchased from Sigma Aldrich and used as received.
8. If crystals are not formed, the following methods can be used to initiate the crystallization: i) scratching the bottom surface of the flask with a metal spatula to initiate the crystallization and storing the reaction mixture at -20 °C for 12 h, ii) adding an extra 10 mL
diethyl ether as antisolvent and storing the reaction mixture at -20 °C for 12 h, iii) concentrating the reaction mixture to dryness, adding
dichloromethane (20 mL) and
diethyl ether (20 mL), and storing at -20 °C for 12 h.
9. Medium porosity sintered glass frits of different size or a Buchner funnel with filter paper (medium-fine porosity) can also be used for vacuum filtration.
10. Authors' procedure: the crystals are crushed into fine powder by the bottom surface of a 24/40 glass stopper to maintain the uniformity of the solid and get rid of the residual solvent from the crystal lattice. A representative process has been shown in Figure 8.
Figure 8. A. Crystals for 2 (reproduced from Figure 5C); B. 24/40 Glass stopper used to grind the crystals into fine powder; C. Fine powder form of 2 before drying under vacuum
11. Vacuum strength of <0.2mm Hg has been used for drying the reported compounds.
12. Checker's second run provided 27.22 g (95%) of product.
13. Light orange crystalline powder (97% purity by quantitative
1H NMR
pdf using
1,3,5-trimethoxybenzene from Sigma Aldrich as internal standard),
1H NMR
pdf (500 MHz, CDCl
3) δ 8.23 (ddd,
J = 4.8, 1.9, 0.8 Hz, 1H), 8.10 (s, 2H), 7.94 - 7.88 (m, 2H), 7.75 (dt,
J = 8.1, 0.9 Hz, 1H), 7.63 - 7.47 (m, 8H), 7.36 - 7.24 (m, 6H), 7.12 (ddd,
J = 7.5, 4.8, 1.0 Hz, 1H).
13C NMR
pdf (126 MHz, CDCl
3) δ 158.4, 156.3, 151.7, 148.2, 139.2, 134.8, 132.6, 132.3, 130.4, 129.94, 129.88, 128.6, 128.6, 126.3, 125.4, 125.3.
19F NMR
pdf (471 MHz, CDCl
3) δ -153.04, -153.09. IR (ATR, cm
-1): 1621, 1594, 1570, 1557, 1498, 1471, 1433, 1412, 1359, 1311, 1252, 1230, 1198, 1152, 1058; HRMS-ESI
+ m/z: [M]
+ calcd. for C
28H
21N
2(+), 385.1699; found, 385.1689.
14. Authors:
ethanol, 200 proof, was purchased from Fisher Scientific and used as received. Checkers:
ethanol, 200 proof, anhydrous, was purchased from Pharmco and used as received.
15.
Hydrazine monohydrate (98+%) was purchased from Thermo Scientific and used as received.
16. The authors used a Vigreux column, other types of reflux condensers such as Liebig condensers can be used with circulating cold air.
17. If crystals are not formed, scratch the bottom surface of the flask with a metal spatula and store the reaction mixture at -20 °C for 12 h.
18. The flask which is used for first recrystallization can be reused for second recrystallization to obtain compound
2.
19. In one instance observed by the checker, the bulk of the second crop of product obtained upon combining the crystallization liquors with the first 50 mL
diethyl ether rinse formed a viscous/waxy oil that coated the sides of the receiving flask. The second 50 mL
diethyl ether cake rinse was collected in this flask (100 mL
diethyl ether in total and initial liquors), the flask capped with a plastic stopper, then the contents allowed to age at room temperature for 24 hr. The following day, the oil solidified to form a brittle solid that coated the sides of the flask. The solids were scraped from the flask using a metal spatula, then isolated via filtration and combined with the solids from the previous filtrations. The combined solids were further rinsed with
diethyl ether (50 mL). No impact on product purity was observed.
20. Off-white crystalline powder (98% purity by quantitative
1H NMR
pdf using
1,3,5-trimethoxybenzene from Sigma Aldrich as internal standard),
1H NMR
pdf (500 MHz, CDCl
3) δ 7.81 (s, 2H), 7.80 - 7.76 (m, 4H), 7.74 - 7.68 (m, 2H), 7.62 - 7.49 (m, 9H), 6.10 (s, 2H).
13C NMR
pdf (126 MHz, CDCl
3) δ 152.8, 152.1, 134.1, 131.9, 131.8, 130.2, 129.9, 129.7, 129.6, 127.8, 125.8.
19F NMR
pdf (471 MHz, CDCl
3) δ -152.78, -152.83.IR (ATR, cm
-1): 3372, 1620, 1600, 1549, 1498, 1446, 1416, 1242, 1199, 1161, 1057; HRMS-ESI
+ m/z: [M]
+ calcd. for C
23H
19N
2(+), 323.1543; found, 323.1537.
21.
Tetrabutylammonium iodide (98%) was purchased from Sigma Aldrich and used as received.
22. Molecular sieves, 4 Å, beads, 1.6-2.5 mm are activated by storing in a 160 °C oven for at least 24 h prior to use.
23. The Schlenk side arm and headspace, the checker inserted an 18-gauge needle through the rubber septum, then purged the headspace through the flask side arm for 5 min under positive flow of dry
N2. The side arm was closed, then a long, 18-gauge needle affixed to the Schlenk line was inserted through the septum with the outlet of the needle positioned just above the solids. Magnetic stirring commenced (50 RPM), then the headspace of the flask purged with dry
N2 through the long needle for 1 h. Upon completion of the
N2 purge, the vent needle and
N2 needle were removed sequentially, then the flask placed under positive
N2 pressure through the Schlenk side arm. The authors ensured inert atmosphere by by evacuating the headspace under vacuum, then refilling with
N2 through the Schlenk side arm three times.
24. Authors:
acetonitrile (ACS grade) was purchased from Fisher Scientific and used as received. Checker:
acetonitrile (anhydrous, 99.8%) was purchased from Sigma Aldrich and used as received.
25. Authors:
styrene (containing 4-
tert-butylcatechol as stabilizer, ≥99%) was purchased from Sigma Aldrich and used as received. Checker:
styrene (99%, stabilized with 10-15 ppm 4-
tert-butylcatechol) was purchased from Alfa Aesar and used as received.
26. The authors prepared
iodosylbenzene using the following procedure. A 250-mL single-necked (24/40) round-bottom flask equipped with a round Teflon-coated stir bar (19.1 mm × 8 mm) is charged with diacetoxyiodobenzene (8.00 g, 97%, 24.8 mmol, 1.00 equiv) which is purchased from Ambeed and used as received. A separate 125-mL Erlenmeyer flask is charged with
Sodium hydroxide pellets (4.96 g, 124 mmol, 5.00 equiv) and distilled water (41 mL).
Sodium hydroxide is purchased from Millipore Sigma and is used as received. The
Sodium hydroxide solution is added to the reaction flask. The resulting suspension is stirred at 23 °C for 2 h. Water (15 mL) is added to the reaction mixture, and the suspension is stirred for additional 1 h (Figure 9A). Solids are isolated by vacuum filtration through a 100-mL Buchner funnel with filter paper (5.5 cm, medium porosity) and are washed with three 20 mL portions of water, followed by three 20 mL portions of chloroform. The resulting solids are transferred to a 40-mL scintillation vial, dried under vacuum (<1 mmHg) at 23 °C for 12 h to remove volatiles, and crushed (
Note 10) to fine powder. At this point,
iodosylbenzene (Figure 9B) (5.00 g, 92%) is characterized by
1H NMR, and is used in procedure C. Off-white powder,
1H NMR (400 MHz, MeOD) δ 8.12-7.95 (m, 2H), 7.65-7.50 (m, 3H). The obtained
1H NMR data agrees with published data.
3 Iodosylbenzene is also commercially available with CAS number 536-80-1. The checker used commercially available
iodosylbenzene (Enamine, 95%).

Figure 9. A. Reaction mixture for the synthesis of iodosylbenzene. B. Isolated iodosylbenzene as fine white powder
27. Celite® 545 was purchased from VWR and used as received.
28. Checker's second run was performed on a 2.5 g scale affording 2.08 g (67%) of product.
29. Off-white crystalline powder (96% purity by quantitative
1H NMR
pdf using
1,3,5-trimethoxybenzene from Sigma Aldrich as internal standard),
1H NMR
pdf (500 MHz, CD
3CN) δ 8.15 (s, 2H), 8.06 - 7.87 (m, 6H), 7.70 - 7.30 (m, 9H), 7.24 - 7.17 (m, 1H), 7.16 - 7.10 (m, 2H), 6.70 - 6.64 (m, 2H), 3.51 (dd,
J = 8.3, 5.7 Hz, 1H), 2.68 (dd,
J = 8.3, 3.1 Hz, 1H), 2.37 (dd,
J = 5.7, 3.1 Hz, 1H).
13C NMR
pdf (126 MHz, CD
3CN) δ 154.3, 154.1, 134.9, 134.8, 132.9, 132.6, 132.5, 130.8, 130.6, 130.0, 129.14, 129.12, 128.8, 127.2, 126.4, 54.5, 49.1.
19F NMR
pdf (471 MHz, CD
3CN) δ -151.76, -151.81. IR (ATR, cm
-1): 1620, 1564, 1497, 1464, 1415, 1248, 1191, 1164, 1054; HRMS-ESI
+ m/z: [M]
+ calcd. for C
31H
25N
2(+), 425.2012; found, 425.2007.
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Aziridines are the smallest nitrogen-containing heterocycles and are important structural motifs in synthetic chemistry because, 1) these scaffolds are present in different natural products and biologically relevant molecules,
4,5,6,7 and 2) they can be used in ring-opening sequence to provide a diverse set of aminofunctionalization products.
7,8 Conventional syntheses of aziridines often provide aziridines bearing electron-withdrawing groups at nitrogen (
e.g.,
N-sulfonyl or
N-carbamoyl aziridines). These aziridines are not compatible with derivatization at nitrogen and structural elaboration requires multi-step deprotection, functionalization sequences.
9,10,11,12,13,14 Progress has been made towards N-H aziridines,
15,16,17,18,19,20 but C-N coupling of these compounds is not well developed.
21,22,23,24 We have developed
N-pyridinium aziridines as synthetic linchpins to access a broad array of
N-functionalized aziridines. Reductive N-N activation can be leveraged both for C-N cross-coupling modalities to access
N-aryl aziridines
25 and to generate
N-aziridinyl radicals, which can engage in olefin addition chemistry.
26 Moreover,
N-pyridinium aziridines participate in ring-opening chemistry to access an array of 1,2-aminofunctionalized products and
β-phenethylamines.
25, 27
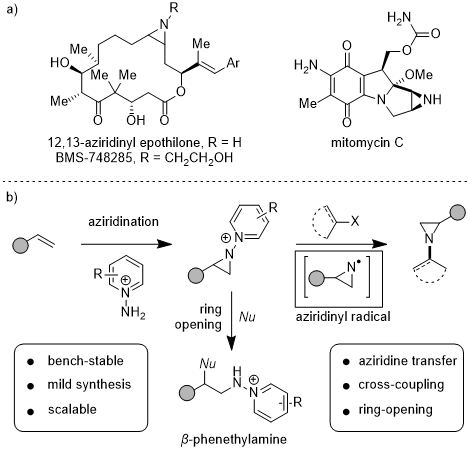
Figure 10. a) Examples of bioactive aziridines. b) Summary of synthesis and reactivity of N-pyridinium aziridines
N-pyridinium aziridines engage in Ni-catalyzed cross-coupling with aryl boronic acids to afford
N-aryl aziridines.
25 Aziridine cross-coupling is productive for a wide variety of aryl boronic acids (
4a-4e) including pharmaceutically derived complex boronic acids such as indomethacin (
4f) and loratadine (
4g).
Ortho-substituted aryl boronic acids are not compatible for this method. Observation of C-N coupling of
N-pyridinium aziridines contrasts with the widely developed Ni-catalyzed ring opening of
N-sulfonyl aziridines with carbon centered nucleophiles via C-N activation.
28,29,30,31,32,33,34,35 We hypothesize that access to C-N coupling reactivity arises due to the low-lying pyridine-centered LUMO: Single-electron transfer from low-valent Ni species would trigger N-N cleavage and provide entry to reactive nitrogen species for coupling.
Figure 11. Ni-catalyzed C-N cross-coupling of N-pyridinium aziridines and aryl boronic acids provides access to diverse N-aryl aziridines
The reductive lability of the N-N bonds of
N-pyridinium aziridines provides a mechanism to generate and utilize
N-aziridinyl radical fragments in synthesis. We demonstrated that reductive photoactivation of
N-pyridinium aziridines (
3, E
onset = -0.85 V
vs. Fc
+/Fc) in presence of Ir(ppy)
3 as photocatalyst (Ir(III)*/Ir(IV) = -1.73 V
vs. Fc
+/Fc) generates
N-aziridinyl radicals which can be transferred to olefinic substrates. When carried out under an O
2 atmosphere, these reactions afford products of olefin 1,2-hydroxyaziridination.
26 Various aziridine precursors including activated and unactivated pyridinium aziridines exhibit moderate to good yields in this transformation. Stability of the benzylic radical is a key factor for an efficient turnover resulting inefficiency of aziridine transfer to unactivated olefins. Regarding the mechanism of aziridine transfer, Stern-Volmer quenching studies indicate that
2 is the primary quencher for this reaction and photogenerated
N-aziridinyl radicals have been trapped by
N-tert-butyl-
α-phenylnitrone (PBN) with its corresponding EPR data to support the
N-aziridinyl radical intermediate. These findings confirm the viability of
N-aziridinyl radicals as reactive intermediates in synthetic chemistry and highlight aziridine group transfer as a feasible synthetic disconnection.
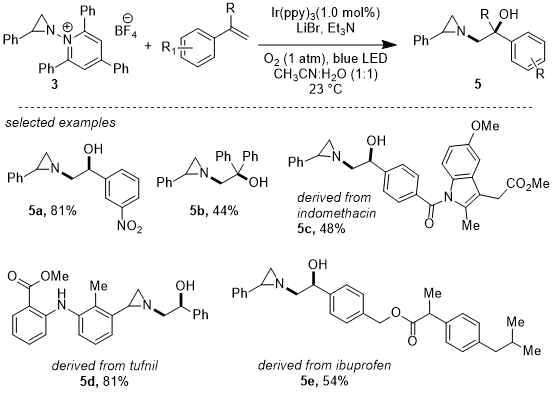
Figure 12. Photochemical 1,2-hydroxyaziridination of olefins via aziridinyl radical transfer from N-pyridinium aziridines
In addition to activation of aziridine functionalization via activation of the N-N linkages of
N-pyridinium aziridines, these substrates also engage in predictable ring-opening chemistry to afford diverse 1,2-aminofunctionalized scaffolds. Lewis acid-mediated ring-opening with halide, hydroxide, alkoxide, amine, and sulfur nucleophiles affords 1,2-aminofunctionalized products (
6a-6d).
25 The products of ring opening with carbon nucleophiles can be achieved in a two-step, one-pot protocol:
N-pyridinium aziridine ring opening with bromide followed by selective Ni-catalyzed Negishi cross-coupling with organozinc nucleophiles affords
β-functionalized phenethylaminopyridinium salts. These products can be further elaborated to
N-aryl,
N-alkyl
β-phenethylamines, as well as products derived from
N-centered radical intermediates, by reductive N-N activation.
27 This method generates pharmaceutically relevant
β-phenethylamines from common
N-pyridinium aziridine starting materials.
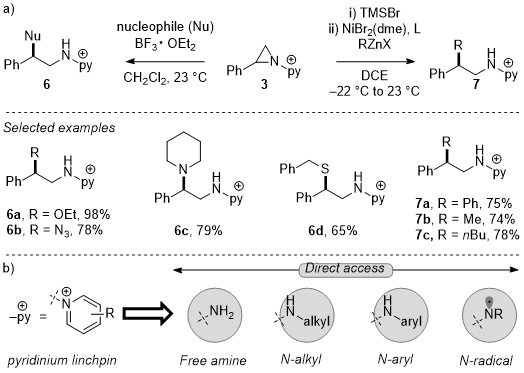
Figure 13. a) Ring-opening chemistry of N-pyridinium aziridines. b) Direct access of diverse β-phenethylamines by derivatization of pyridinium linchpins
In summary, N-pyridinium aziridines expand the synthetic chemistry of aziridine scaffolds by enabling both rapid structural diversification and novel synthetic disconnections. Hence, reliable syntheses of N-pyridinium aziridines 3 is essential to advance the synthesis and application of this functional group.
Appendix
Chemical Abstracts Nomenclature (Registry Number)
2,4,6-Triphenylpyrylium tetrafluoroborate; (448-61-3)
2-Aminopyridine; (504-29-0)
Hydrazine monohydrate; (7803-57-8)
Tetrabutylammonium iodide; (311-28-4)
4 Å molecular sieves; (70955-01-0)
Styrene; (100-42-5)
Iodosylbenzene; (536-80-1)

|
Samya Samanta obtained his B.Sc. and M.Sc. degrees in Chemistry at the Indian Institute of Technology, Kharagpur (India) under the supervision of Prof. N. D. Pradeep Singh. During his undergraduate studies, he also worked as a research intern under Prof. David Berg at University of Victoria (Canada) and under Prof. Leong Weng Kee at Nanyang Technological University (Singapore). Currently his research is on amine transfer reagent design under Prof. David C. Powers at Texas A&M University (USA). |

|
Hao Tan obtained his B.Sc. degree in Chemistry at Nankai University, Tianjin (China), under the supervision of Prof. Zhengjie He. He also worked as a visiting undergraduate researcher under Prof. Ohyun Kwon at University of California, Los Angeles (USA). During his undergraduate studies, his research focused on phosphine catalysis. Currently, his research is on aziridination using N-aminopyridinium reagents under Prof. David C. Powers at Texas A&M University (USA). |

|
Saikat Chatterjee received his B.Sc. in Chemistry from the University of Burdwan and his M.Sc. from Banaras Hindu University, where he conducted research under the supervision of Prof. K. N. Singh. He obtained his Ph.D. from the Indian Institute of Science Education and Research (IISER) Bhopal under the mentorship of Prof. Sreenivas Katukojvala, working on Rh-catalyzed and visible-light mediated carbene-transfer reactions. He is currently a postdoctoral researcher in the laboratory of Prof. David C. Powers at Texas A&M University, where his research focuses on non-aqueous redox flow batteries and the synthetic chemistry of N-aminopyridinium reagents. |

|
David C. Powers received his undergraduate education at Franklin and Marshall College where he pursued undergraduate research with Prof. Phyllis Leber. He obtained a Ph.D. from Harvard University in 2012 working with Prof. Tobias Ritter and pursued post-doctoral training with Prof. Daniel Nocera at the Massachusetts Institute of Technology and Harvard University. In 2015, he was appointed as Assistant Professor in the Department of Chemistry at Texas A&M University where he was promoted to Associate Professor in 2021 and Professor in 2023. His research program utilizes tools of organic, inorganic, and in crystallo chemistry to advance sustainable synthetic chemistry. |

|
Michael M. Gilbert received his undergraduate degree at the University of Massachusetts-Amherst where he pursued undergraduate research with Prof. Vincent Rotello. He obtained a Ph.D. from the University of Michigan working with Prof. John Montgomery and pursued post-doctoral training with Prof. Dan Weix at the University of Wisconsin Madison. In 2022, he began his industrial career as a Senior Scientist in Process Chemistry at AbbVie. |

|
Nathan D. Ide received his undergraduate degree from Hope College, where he pursued undergraduate research with Prof. Stephen K. Taylor. He obtained a Ph.D. from the University of Illinois at Urbana-Champaign, working under the guidance of Prof. David Y. Gin. He currently serves as the Head of Process Chemistry at AbbVie. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved