Org. Synth. 2026, 103, 101-120
DOI: 10.15227/orgsyn.103.0101
Synthesis of Aryl Halides via a Safer Sandmeyer Reaction Protocol
Submitting Authors: Tim Schulte, Felix Waldbach, Tobias Ritter,
1*
Checking Authors: Kunj Kapur, Daniya Aynetdinova, and Nuno Maulide*
21. Procedure (Note 1)
A. 4-Chlorobenzonitrile (1). Under an ambient atmosphere, to a 250-mL round-bottomed flask (29/32) equipped with a Teflon-coated magnetic stir bar (oval shape, 30 mm × 15 mm) (Fig. 1A) was added 4-aminobenzonitrile (3.54 g, 30.0 mmol, 1.0 equiv) (Note 2), copper(I) chloride (2.97 g, 30.0 mmol, 1.0 equiv) (Note 3), and benzyltrimethylammonium chloride (5.57 g, 30.0 mmol, 1.0 equiv) (Note 4). Then, acetonitrile (75 mL, c = 0.40 M) (Note 5) was added using a 100-mL measuring cylinder, followed by 2-ethylhexyl nitrate (10.9 mL, 10.5 g, 60.0 mmol, 2.0 equiv) (Note 6), which was added in one portion via a 20 mL plastic syringe with a stainless steel needle (0.8 × 120 mm, G21 fitting). Subsequently, 2-methyl-2-butene (1.6 mL, 1.1 g, 15 mmol, 0.5 equiv) (Note 7) was added in one portion via a 2-mL plastic syringe with a stainless steel needle (0.8 × 120 mm, G21 fitting) (Fig. 1B). A Dimroth condenser (29/32, 330 mm) was attached to the round-bottomed flask and secured with a stainless steel clamp, then cold water was circulated through the condenser. The flask was placed in an oil bath preheated to 85 °C, and the reaction mixture was stirred for 16 h at 600 rpm in the 85 °C oil bath (Notes 8 and 9).

Figure 1. A. Reaction set up. B. Appearance of the reaction mixture after addition of all chemicals. C. Appearance of the reaction mixture after heating at 85 °C for 16 h, and subsequently cooling to 23 °C (photos provided by the authors).
After cooling to 23 °C (over a period of 45 min after removal of the oil bath) (Fig. 1C), the reaction mixture was diluted with ethyl acetate (75 mL) (Note 10) and transferred to a 250-mL separatory funnel. A further 30 mL EtOAc was used to transfer the remaining residue from the flask (Note 11, Fig. 2A). A saturated sodium thiosulfate solution was prepared by adding sodium thiosulfate pentahydrate (370 g) (Note 12) to 300 mL of deionized water. The organic layer was washed with the prepared sodium thiosulfate solution (4 × 75 mL) to remove any reactive chlorine species (Fig. 2B). The organic phase was transferred to a 500-mL beaker. The aqueous phases were combined and extracted with ethyl acetate (2 × 75 mL). The organic layers were combined and then dried over 15 g of anhydrous MgSO4 (Note 13). The mixture was then filtered through fluted filter paper (Whatman Cytiva, 100 mm) in a glass funnel into a 500-mL round-bottomed flask (29/32) (Fig. 2C).

Figure 2. A. Reaction mixture partitioned between ethyl acetate and the first portion of saturated sodium thiosulfate solution in a separatory funnel; B. Mixture after third wash with saturated sodium thiosulfate solution; C. Solution after drying with MgSO4 and filtration (photos provided by the checkers).
Silica gel (30 g) (Note 14) was added to the filtrate and the solvent was removed under reduced pressure on a rotary evaporator (40 °C, 120 rpm, 150 mbar for 15 min, then 40 °C, 120 rpm, 120 mbar for 15 min, followed by 40 °C, 120 rpm, 15 mbar for 30 min) (Note 15). Purification was carried out by flash column chromatography on silica gel (200 g) (Note 14) using a glass column (60 x 450 mm) and a solvent mixture of ethyl acetate in pentane (5% V/V, 2500 mL) (Note 16, Fig. 3A) as eluent (Note 17). Fractions of 50 mL volumes were collected and analyzed by TLC (Note 18, Fig. 3B). The fractions containing product 1 were pooled (Note 17), and the solvent was removed under reduced pressure using a rotary evaporator (40 °C, 150 rpm, 600 mbar for 10 min, followed by 40 °C, 150 rpm, 150 mbar for 25 min) followed by high-vacuum (23 °C, <1×10-2 mbar for 16 h) (Note 19), to give the desired 4-chlorobenzonitrile (1) as a colorless solid (Note 20) in 65% yield (2.68 g, 19.5 mmol) and 100 % purity as determined by qNMR with diethyl malonate as the internal standard (Notes 21 and 22, Fig. 3C).

Figure 3. A. Column set up with dry load and solvent reservoir. B. TLC of crude mixture with ethyl acetate in pentane (5% V/V) as eluent, Rf of compound 1 = 0.49; C. Product after drying (photos A and C provided by the authors; photo B provided by checkers).
B. 1-Bromo-2-methyl-3-nitrobenzene (2). Under an ambient atmosphere, to a 250-mL round-bottomed flask (29/32) equipped with a Teflon-coated magnetic stir bar (oval shape, 30 × 15 mm) (Fig. 4A) was added 2-methyl-3-nitroaniline (3.04 g, 20.0 mmol, 1.0 equiv) (Note 23), tetrabutylammonium nitrate (12.2 g, 40.0 mmol, 2.0 equiv) (Note 24), copper(I) bromide (574 mg, 4.00 mmol, 0.2 equiv) (Note 25), and sodium thiosulfate pentahydrate (993 mg, 4.00 mmol, 0.2 equiv) (Note 12). Subsequently, acetonitrile (50 mL, c = 0.4 M) (Note 5) was added using a 50-mL measuring cylinder, followed by 1,2-dibromoethane (5.2 mL, 11 g, 60 mmol, 3.0 equiv) (Note 26), which was added in one portion using a 10 mL plastic syringe with a stainless steel needle (0.8 × 120 mm, G21 fitting). A Dimroth condenser (29/32, 290 mm) was attached to the round-bottomed flask and secured with a stainless steel clamp. Cold water was circulated through the condenser. The flask was placed in an oil bath preheated to 85 °C, and the reaction mixture was stirred for 16 h at 600 rpm in the 85 °C oil bath (Notes 8 and 9) (Fig. 4B).

Figure 4. A. Reaction set up. B. Appearance of the reaction mixture after addition of all chemicals and at the beginning of the heating period. C. Appearance of the reaction mixture after heating at 85 °C for 16 h, and subsequently cooling to 23 °C (photos provided by the authors).
After cooling to 23 °C (over a period of 45 min after removal of the oil bath) (Fig. 4C), the reaction mixture was diluted with ethyl acetate (50 mL) (Note 10) and transferred to a 250-mL separatory funnel. A further 30 mL EtOAc was used to transfer the remaining residue in the flask (Note 11, Fig. 5A). A saturated sodium thiosulfate solution was prepared by adding sodium thiosulfate pentahydrate (370 g) (Note 12) to 300 mL of deionized water. The organic layer was washed with the prepared sodium thiosulfate solution (4 × 50 mL) (Fig. 5B) to remove any reactive bromine species. The organic phase was transferred to a 500 mL beaker and the combined aqueous phases were extracted with ethyl acetate (2 × 50 mL). The combined organic layers were then dried over 10 g of anhydrous Na2SO4 (Note 27), filtered through fluted filter paper (Whatman Cytiva, 100 mm) in a glass funnel into a 500 mL round-bottomed flask (29/32) (Fig. 5C).

Figure 5. A. Reaction mixture diluted with ethyl acetate and transferred to a separatory funnel. B. Mixture after third wash with saturated sodium thiosulfate solution C. Solution after drying with anhydrousNa2SO4 and filtration (photos provided by the authors).
Silica gel (32 g) (Note 14) was added to the filtrate, and the solvent was removed under reduced pressure on a rotary evaporator (40 °C, 120 rpm, 150 mbar for 15 min, followed by 40 °C, 120 rpm, 15 mbar for 15 min) (Note 15, Fig. 6B). Purification was carried out by flash column chromatography on silica gel (200 g) (Note 14) using a glass column (70 x 540 mm) and a solvent mixture of ethyl acetate in pentane (1%, V/V, 3500 mL) (Note 16, Fig. 6C) as eluent (Note 28). Fractions of 50 mL volumes were collected and analyzed by TLC (Note 18, Fig. 6A). The fractions containing product 2 (fractions 37-57) were combined (Note 28). The solvent was removed under reduced pressure using a rotary evaporator (40 °C, 150 rpm, 600 mbar for 20 min, followed by 40 °C, 150 rpm, 150 mbar for 30 min), followed by high-vacuum (23 °C, <1×10-2 mbar for 16 h) (Note 19), to give the desired 1-bromo-2-methyl-3-nitrobenzene (2) as a colorless solid (Note 29) in 67% yield (2.89 g, 13.4 mmol) and 98.6% purity as determined by qNMR with diethyl malonate as the internal standard (Notes 30 and 31, Fig. 6D).

Figure 6. A. TLC of crude mixture with ethyl acetate in pentane (5% V/V) as eluent, Rf of compound 2 = 0.86; B. Dry load. C. Column set up with dry load and eluent. D. Purified product 2 after drying (photo A provided by the checkers, photos B-D provided by the authors).
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/about/governance/committees/chemical-safety.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
4-aminobenzonitrile,
copper(I) chloride,
benzyltrimethylammonium chloride,
2-ethyl-1-hexyl nitrate,
2-methyl-2-butene,
acetonitrile,
acetone,
pentane, silica gel,
MgSO4,
Na2SO4,
sodium thiosulfate pentahydrate,
2-methyl-3-nitroaniline,
tetrabutylammoniumnitrate,
1,2-dibromoethane, and
copper(I)bromide.
2.
4-Aminobenzonitrile (98%) was purchased from BLD Pharmatech, and used as received.
3.
Copper(I) chloride (>98%) was purchased from TCI and used as received.
4.
Benzyltrimethylammonium chloride (>99.0%) was purchased from TCI and used as received. Tetrabutylammonium chloride can also be used. In the absence of an additional chloride source.
25.
Acetonitrile (>99.9%, HPLC grade) was purchased from Fisher Scientific and used as received.
6.
2-Ethylhexyl nitrate (>97.0%) was purchased from Santa Cruz Biotechnology and used as received.
2-Ethylhexyl nitrate was used because it is an inexpensive, commercially available nitrate ester, but other nitrate esters can also be used. Fewer equivalents of the reagent resulted in lower yields.
37.
2-Methyl-2-butene (>99%) was purchased from Fisher Scientific and used as received. It was used as a scavenger for reactive chlorine species formed during the reaction.
38. The stirring plate was purchased from Heidolph Instruments GmbH & Co. KG, with an output power of 230-240 V (50-60 Hz, 825 W), and a stirring rate of 100-1400 rpm.
9. The silicone oil used in the oil bath was purchased from ABCR GmbH & Co. KG, and has a boiling point of >205 °C. The level of the oil should be approximately the same height as the level of the reaction mixture, after reaching the maximum reaction temperature. The temperature was monitored with the temperature probe of the heating-stir plate. The temperature has a significant impact on reaction yields. In a run performed by the checkers, a malfunction of the heating plate led to a mismatch between the temperature probe reading and the actual oil bath temperature (around 30 °C discrepancy), which afforded substantially lower yields.
10.
Ethyl acetate (≥99.0%) was purchased from VWR, and distilled prior to use.
11. The mixture was transferred to the separatory funnel by first decanting the suspension from the flask
via a glass funnel into the separatory funnel, and subsequently rinsing the flask with
ethyl acetate (3 × 10 mL).
12.
Sodium thiosulfate pentahydrate (>99.5%) was purchased from Thermo Scientific and used as received. Notably, the
sodium thiosulfate pentahydrate takes around 50 min to dissolve completely. Using fewer equivalents of the reagent resulted in lower yields.
13. Anhydrous
magnesium sulfate (99.0%) was purchased from Thermo Scientific, and used as received. After adding the
MgSO4, a Teflon-coated magnetic stir bar (rod shape, 40 mm × 7 mm) was also added, and the combined organics were stirred at 400 rpm at 23 °C for 10 min.
14. Silica gel (Silica gel 60, pore size 40-63 μm) was purchased from VWR and used as received.
15. A Büchi R-210 Rotavapor was used in combination with a Büchi V-850 controller and a Büchi V-700 vacuum pump. Water was used as the heating bath medium, and both pressure and temperature were monitored directly
via the controller.
16.
Pentane (>95.0%) was purchased from OQEMA GmbH, and distilled before use.
17. Flash column chromatography was carried out as follows: A glass-column (60 × 450 mm), with a 1000 mL reservoir was charged with 200 g
SiO2 (Silica gel 60 with pore size 40-63 μm) and 350 mL
ethyl acetate in
pentane (5% V/V). Crude compound
1 (previously adsorbed onto 30 g of silica gel) was applied on the top of the column, followed by sand (50 g). The column was eluted using 2500 mL
ethyl acetate in
pentane (5%, V/V). Fractions of 50 mL volumes were collected. TLC analysis of the fractions was done with
ethyl acetate in
pentane (1% V/V) as eluent and visualized with 254-810 nm UV light. Product
1 was identified in fractions 17-25.
Figure 7. TLC plates for the column chromatography of 1 developed with ethyl acetate in pentane (5% V/V) as eluent, collected in 50 mL fractions. The product was found in fractions 17-25 (photos provided by the checkers).
18. Thin layer chromatography (TLC) was performed using EMD TLC plates (silica gel 60 F254 pre-coated at 250 μm thickness) and visualized by fluorescence quenching under 254 nm UV light.
19. High vacuum corresponds to a vacuum below 1×10
-2 mbar. The vacuum pump was purchased from Vacuubrand GmbH & Co. KG.
20. Compound
1 is characterized as follows: colorless solid; mp: 89.3-89.8 °C;
1H NMR
pdf (500 MHz, CDCl
3, 23°C, δ): 7.67-7.55 (m, 2H), 7.54-7.36 (m, 2H) ppm;
13C NMR
pdf (126 MHz, CDCl
3, 23°C, δ): 139.7, 133.5, 129.8, 118.1, 110.9 ppm; HRMS-EI(m/z) calc'd for C
7H
4N
35Cl
+ [M]
+, 137.0027; found, 137.0029; deviation: +1.4 ppm. HRMS-EI(m/z) calcd for C
7H
4N
37Cl
+ [M]
+, 138,9998; found, 138.9995, deviation: -2.2 ppm. Compound
1 is bench stable.
21. Quantitative
1H NMR
pdf spectroscopy (500 MHz, CDCl
3) was performed using diethylmalonate (TCI, >99.0% purity, used as received) as internal standard. The NMR sample was prepared by weighing 23.7 mg of the internal standard and 30.7 mg of
1 into a 4 mL borosilicate vial. Subsequently, 1.0 mL CDCl
3 was added and the mixture was vortexed for 30 seconds to fully dissolve all solids. Then, a 0.5 mL aliquot was transferred to a 5 mm NMR tube. A
1H NMR spectrum was recorded with the following parameters: scan number: 16, D1: 35 sec, spectra offset: 6.175 ppm (3089.62) Hz, spectra width: 19.988 ppm. The integration of the
1H signals showed a molar ratio of 1.505, corresponding to 100 wt% of
4-chlorobenzonitrile (
1).
22. The reaction was performed by the checkers twice on full scale (30.0 mmol) and once on half scale (15.0 mmol). Reaction yields for the other two attempts: Full scale (Run 2, 30 mmol): 69% (2.83 g), 97.7% purity. Half scale (Run 3, 15 mmol): 60% (1.24 g), 100% purity.
23.
2-Methyl-3-nitroaniline (98%) was purchased from BLD Pharmatech and used as received.
24.
Tetrabutylammonium nitrate (98.0%) was purchased from Fisher Scientific and used as received. Using fewer equivalents of the reagent resulted in lower yields.
25.
Copper(I) bromide (>97%) was purchased from Fluorochem, and used as received.
26.
1,2-Dibromoethane (>98.0%) was purchased from Sigma Aldrich, and used as received. Using fewer equivalents of the reagent resulted in lower yields.
27. Anhydrous
Na2SO4 (>98.5%) was purchased from VWR and used as received. After adding the
Na2SO4, a Teflon-coated magnetic stir bar (rod shape, 40 mm × 7 mm) was also added, and the combined organics were stirred at 400 rpm at 23 °C for 10 min.
28. Flash column chromatography: A glass-column (70 mm × 540 mm, with a 1000 mL reservoir) was charged with 200 g
SiO2 (Silica gel 60 with pore size 40-63 μm) and 400 mL
ethyl acetate in
pentane (1% V/V). Crude compound
2 (previously adsorbed onto 32 g of silica gel) was applied on the top of the column, followed by sand (40 g). The column was eluted with 3500 mL
ethyl acetate in
pentane (1%, V/V). Fractions of 50 mL volumes were collected. TLC analysis of the fractions was done with
ethyl acetate in
pentane (5% V/V) as eluent and visualized with 254 nm UV light. Product
2 was identified in fractions 37-57.
Figure 8. TLC plates for the column chromatography of compound 2 developed with ethyl acetate in pentane (5% V/V) as eluent. Fraction volume: 20 mL. For the half-scale reaction, fractions 42-64 contained product 2 and were pooled (photos provided by the checkers).
29. Compound
2 is characterized as follows: colorless solid; mp: 38.0-38.8 °C;
1H NMR
pdf (500 MHz, CDCl
3, 23°C): δ 7.78 (dd,
J = 8.1, 2.9 Hz, 1H), 7.71 (dd,
J = 8.1, 2.9 Hz, 1H), 7.23-7.16 (m, 1H), 2.57 (s, 3H) ppm;
13C NMR
pdf (126 MHz, CDCl
3, 23°C): δ 151.5, 136.9, 132.5, 127.6, 127.4, 123.2, 19.4 ppm; HRMS-EI(m/z) calc'd for C
7H
6NO
279Br
+ [M]
+, 214.9577; found, 214.9585; deviation: +3.7 ppm. HRMS-EI(m/z) calc'd for C
7H
6NO
281Br
+ [M]
+, 216.9556; found, 216.9564; deviation: +3.7 ppm. The compound is bench stable.
30. Quantitative
1H NMR
pdf spectroscopy (500 MHz, CDCl
3) was performed using diethylmalonate (TCI, >99% purity, used as received) as internal standard. The NMR sample was prepared by weighing 26.8 mg of the internal standard and 14.0 mg of
2 into a 4 mL borosilicate vial. Subsequently, 1.0 mL CDCl
3 was added and the mixture was vortexed for 30 seconds to fully dissolve all solids. Then, a 0.5 mL aliquot was transferred to a 5 mm NMR tube. A
1H NMR spectrum was recorded with the following parameters: scan number: 16, D1: 35 sec, spectra offset: 6.175 ppm (3089.62) Hz, spectra width: 19.988 ppm. The integration of the
1H signals showed a molar ratio of 0.386, corresponding to 98.6 wt% of
1-bromo-2-methyl-3-nitrobenzene (
2).
31. The reaction was performed by the checkers once on full scale (Run 1, 30.0 mmol) and once on half scale (Run 2, 15.0 mmol). The half scale (15.0 mmol) run provided
2 in 66% yield (1.43 g) and 98.1% purity.
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Since their discovery in the 19
th century,
4 diazonium salts are commonly used in organic synthesis on both industrial and laboratory scale,
5 due to their high reactivity when compared to other aryl(pseudo)halides.
3 The Sandmeyer halogenation represents one of the earliest applications for diazonium salts and is still used today.
6 Formation of dinitrogen contributes to the thermodynamic driving force for the desirable reaction but is also reason for safety concerns due to the unpredictable stability of the diazonium compounds.
7 Accidents have occurred during the preparation or conversion of diazonium salts, including Sandmeyer reactions.
8 Our group has recently demonstrated a safer one-step diazotization-halogenation protocol that is based on rate-limiting nitrate reduction.
3 In contrast to the traditional preparation method for diazoniums, which is based on nitrite reagents,
3 less expensive nitrate reagents can be used in our protocol. Because nitrate is kinetically more stable than nitrite, it can coexist with a variety of nucleophiles that are oxidized by nitrite. In situ and rate-determining reduction of nitrate therefore allows to access aryldiazoniums as fleeting intermediates only, with immediate conversion that avoids their dangerous accumulation.
3 Deaminative iodination has been achieved using SO
2 as a chemically competent reductant for nitrate.
3 The low solubility of SO
2 at the reaction temperatures
9 requires closed reaction vessels. Deaminative bromination and chlorination can be carried out at reflux, open to the atmosphere. For the deaminative bromination, tetrabutylammonium nitrate is used. The use of less expensive nitrate salts (such as KNO
3) or the combination of KNO
3 with phase transfer catalysts, result in lower yields (Fig. 9).
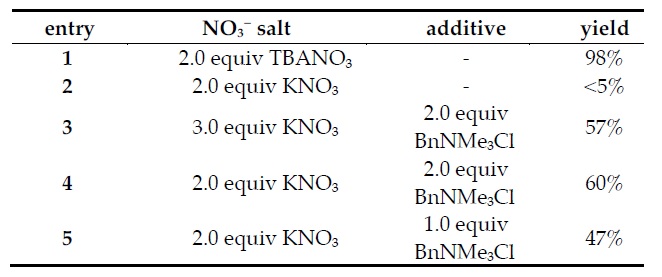
Figure 9. Yields for deaminative bromination with different nitrate salts and in the presence of phase transfer catalysts.
When the amount of 1,2-dibromoethane is decreased from 3.0 equiv to 2.0 equiv or 1.0 equiv in case of
2, similar yields are observed (Fig. 10). However, for other substrates, lower yields were obtained when the amount of 1,2-dibromoethane was decreased below 3.0 equiv.
3Figure 10. Yields for deaminative bromination with different amounts of 1,2-DBE.
The deaminative chlorination is carried out with stoichiometric CuCl. In contrast to the deaminative bromination, substoichiometric amounts of CuCl result in lower yield of the deaminative chlorination (Fig. 11).
10 Also the combination of catalytic amounts of CuCl with ligands, or the combination of CuCl with CuCl
2, which has previously enabled the use of catalytic copper in Sandmeyer reactions,
11 did not lead to a significant increase in yield.
Figure 11. Yields for deaminative chlorination with different Cu loadings and in the presence of ligands.
10The use of an additional equivalents of ammonium chloride salt leads to higher yields of the deaminative chlorination. In the original report,
3 tetrabutylammonium chloride was used. In the presence of the more inexpensive BnNMe
3Cl, the reaction proceeds equally well (Fig. 12).
Figure 12. Yields for deaminative chlorination with additives.
Decrease of the nitrate ester loading to 1.5 equivalents resulted in similar yields in case of
2 (Figure 13).
10 However, 2.0 equivalents gave higher yields for a broader range of substrates. If the amount of nitrate ester is further decreased, lower yields are observed.
Figure 13. Yields for deaminative chlorination with different loadings of nitrate ester.
10The deaminative halogenations proceed on a variety of electronically diverse anilines, including complex molecules and amino heterocycles.
3 Other deaminative transformations that are based on the in situ generation of diazonium salts
via nitrate reduction have been reported.
12-13
Appendix
Chemical Abstracts Nomenclature (Registry Number)
4-Aminobenzonitrile: 4-aminobenzonitrile; (873-74-5)
CuCl: copper(I)-chloride; (7758-89-6)
BnNMe3Cl: benzyltrimethylammonium chloride; (56-93-9)
2-Ethylhexyl nitrate: 2-ethylhexyl nitrate; (27247-96-7)
2-Methyl-2-butene: 2-methyl-2-butene; (513-35-9)
2-Methyl-3-nitroaniline: 2-methyl-3-nitroaniline; (603-83-8)
CuBr: copper(I)-bromide (7787-70-4)
Na2S2O3∙5H2O: sodium thiosulfate pentahydrate; (10102-17-7)
1,2-Dibromoethane: 1,2-dibromoethane (106-93-4)

|
Tim Schulte received his undergraduate education at the University of Cologne. He conducted research at the University of Auckland with Prof. Margaret Brimble and at Bayer AG in Wuppertal. He earned his M. Sc. degree with Prof. Hans G. Schmalz and Prof. Tobias Ritter. He obtained his Ph.D. with Prof. Tobias Ritter at Max-Planck-Institut für Kohlenforschung in 2024, and is currently a postdoctoral researcher in the group of Prof. Ritter. |

|
Felix Waldbach completed his apprenticeship as a technician in chemistry in 2019 (Max-Planck-Institut fuer Kohlenforschung, Muelheim an der Ruhr). During this time, he worked in the group of Prof. Benjamin List (homogeneous catalysis, Klussmann) and group of Dr. Nils Theyssen (Technikum). Since 2022 he is working in the group of Prof. Tobias Ritter on organic synthesis and organometallic chemistry. |

|
Tobias Ritter received his undergraduate education in Braunschweig, Bordeaux, Lausanne, and Stanford. He has performed undergraduate research with Prof. Barry M. Trost, obtained his Ph.D. with Prof. Erick M. Carreira at ETH Zurich in 2004, and was a postdoc with Prof. Robert H. Grubbs at Caltech. In 2006, Tobias was appointed as Assistant Professor in the Department of Chemistry and Chemical Biology at Harvard, promoted to Associate Professor in 2010, and to Professor of Chemistry and Chemical Biology in 2012. Since 2015 he is director at the Max-Planck-Institut fuer Kohlenforschung. |

|
Kunj Kapur studied chemistry at the University of Durham in England, undertaking her Masters research in the Medicinal Chemistry department at F. Hoffmann La-Roche in Switzerland. Currently, she is a PhD student at the University of Vienna, under the supervision of Prof. Nuno Maulide. She is conducting research on the deprotective functionalization of sulfonamide-protected compounds. |

|
Daniya Aynetdinova obtained her MChem degree at the University of Oxford in England, undertaking her Masters project in the group of Prof. Timothy J. Donohoe. She subsequently pursued her DPhil as part of the Synthesis for Biology and Medicine CDT Program, carrying out research under the supervision of Prof. Timothy J. Donohoe. Currently, she is a postdoctoral researcher at the University of Vienna in the group of Prof. Nuno Maulide. |

|
Nuno Maulide obtained his Ph.D. at the Université catholique de Louvain under the supervision of Prof. István E. Markó in 2007, followed by a postdoctoral stay in the group of Prof. Barry M. Trost at Stanford University. In 2009, he started his independent career at the Max-Planck Institut für Kohlenforschung. Following this, in 2013, he became a Full Professor at the University of Vienna. His research interests are broadly spread in the field of organic chemistry and include method development, total synthesis and medicinal chemistry-driven investigations. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved