Org. Synth. 2026, 103, 154-170
DOI: 10.15227/orgsyn.103.0154
Preparation of 3-(Diphenylphosphino)propanol (ProPhos) and its Use in Nickel-Catalyzed Suzuki-Miyaura Coupling
Submitting Authors: Jin Yang, Rui Zhang, Tianning Diao*
1Checking Authors: Benjamin S. Y. Chiu and Sophie A. L. Rousseaux
*21. Procedure (Note 1)
A. 3-(Diphenylphosphino)propanol (3). A two-necked 100-mL round-bottomed flask (24/40 joint) equipped with a rubber septum, L-shaped adaptor, and a 3-cm egg-shaped Teflon-coated stir bar is connected to a Schlenk line vacuum manifold and flame-dried. After filling with nitrogen, the reaction flask is cooled to 22 ℃ (Figure 1A). 3-Chloro-1-propanol (2) (1.34 mL, 16.1 mmol, 1.0 equiv) (Note 2) is added to the flask using a 2-mL plastic syringe equipped with a 22-gauge needle, followed by 30 mL of dry THF (Note 3) added using a 50-mL plastic syringe equipped with a 16-gauge needle. The flask was cooled in an ice-water bath. Diphenylphosphine (1) (2.80 mL, 16.1 mmol, 1.0 equiv) (Note 4) is added using a 5-mL plastic syringe equipped with a 22-gauge needle at 0 ℃ (Figure 1B), followed by a solution of n-butyllithium in hexanes (14.2 mL, 35.5 mmol, 2.2 equiv) (Note 5) dropwise over 10 min, using a 20-mL plastic syringe equipped with a 20-gauge needle (Figure 1C) (Note 6).

Figure 1. A. Set-up of flask for the reaction; B. Addition of PPh2H to the THF solution; C. Dropwise addition of n-BuLi to the mixture; D. Reaction mixture after stirring for 1 h at 22 ℃. (Photos provided by the submitting authors)
The rubber septum is sealed with electrical tape, and the flask is removed from the ice-water bath and allowed to warm to 22 ℃. The resulting orange reaction mixture is stirred at 500 rpm for 1 h (Figure 1D). An aliquot of the orange solution is drawn by syringe and checked by TLC analysis (Note 7) and 31P{1H} NMR spectroscopy. After the reaction is complete, deoxygenated NH4Cl (aq.) (40 mL) (Note 8) is added to the reaction mixture by syringe. The mixture is transferred to a 250-mL separatory funnel and extracted with 30 mL of ethyl acetate (Note 9) three times. The organic layers (totaling 130 mL) are combined in a 250-mL Erlenmeyer flask, dried over anhydrous sodium sulfate (15 g) (Note 10), and filtered through a 150-mL filter funnel (glass frit, medium porosity) into a 500-mL round-bottomed flask. The contents of the filter funnel are washed with ethyl acetate (3 x 10 mL), and the combined organics are subjected to rotary evaporation (with a 40 ℃ water bath, 75 mmHg) to remove the volatiles.

Figure 2. A. Column loaded with crude material; B. Isolated ProPhos. (Photos provided by the submitting authors)
Using a glass column (6 cm in diameter x 18 cm in height), a silica gel column is prepared using 140 g of silica gel (Note 11), primed with 500 mL of hexanes (Note 12). The crude product is dissolved in 5 mL of methylene chloride (Note 13) and loaded to the column. The flask containing the residual crude material is rinsed twice with 3 mL of methylene chloride, and these rinsings were also added to the column. A 5 cm layer of sand is placed on top of the column (Figure 2A). Initially, the column is eluted with 10:1 hexanes:ethyl acetate, and 750 mL of eluent is collected to remove any unreacted PPh2H. Subsequently, the column is eluted with 4:1 hexanes:ethyl acetate, and the eluent fractions containing the product (total of 600 mL) (Note 14) are pooled then concentrated on a rotary evaporator under reduced pressure (with a 40 ℃ water bath, from 350 mmHg to 75 mmHg) (Note 15). The resulting oil is further dried under high vacuum (<10 mmHg) overnight, yielding 3-(diphenylphosphino)propanol (ProPhos) (3) (3.71 g, 94%) as a white solid (Figure 2B) (Notes 16 and 17).
B. 4-Acetylbiphenyl (6). Inside a nitrogen-filled glovebox, an oven-dried 20-mL vial equipped with a 1 cm x 0.4 cm egg-shaped stir bar is charged with bis(1,5-cyclooctadiene)nickel(0) (Ni(cod)2) (35.0 mg, 0.13 mmol, 1.0 mol%) (Notes 19 and 20) and ProPhos (3) (123.0 mg, 0.50 mmol, 4.0 mol%). To this mixture, 2 mL of 2-methyltetrahydrofuran (2-MeTHF) (Note 21) is added using a 5-mL plastic syringe equipped with a 22-gauge needle and the reaction mixture was stirred for 30 min at 22 ℃ to afford a deep red catalyst stock solution (Figure 3A). Separately, a 100-mL round-bottomed Schlenk flask with a single-necked 24/40 joint is equipped with a rubber septum and a 3 cm egg-shaped Teflon-coated magnetic stir bar. The Schlenk flask is connected to a Schlenk line vacuum manifold and flame-dried under reduced pressure, and after filling with nitrogen, allowed to cool to 22 ℃. The flask is then charged with 4'-bromoacetophenone (4) (2.50 g, 12.6 mmol, 1.0 equiv) (Note 22), phenylboronic acid (5) (2.30 g, 18.9 mmol, 1.5 equiv) (Note 23), and K3PO4 (6.67 g, 31.4 mmol, 2.5 equiv) (Note 24). The Schlenk flask is brought into the glovebox (Figure 3B) and sealed with a rubber septum before addition of 2-MeTHF (6 mL) using a 10-mL plastic syringe equipped with a 22-gauge needle. This is followed by the addition of the (ProPhos)Ni catalyst solution by syringe (2 mL) (Note 25). The Schlenk flask is sealed, removed from the glovebox, and deoxygenated water (5.3 mL) is added (Note 26) (Figure 3C). The flask is placed in a pre-heated 70 ℃ mineral oil bath for 16 h (700 rpm) under positive nitrogen pressure.

Figure 3. A. Appearance of precatalyst solution; B. Reaction set-up in a nitrogen-filled glovebox; C. Addition of deoxygenated water to the reaction mixture outside the glovebox; D. Reaction mixture after 16 h at 70 ℃. (Photo A provided by submitting authors; photos B-D provided by checking authors)
After heating for 16 h, an aliquot of the yellow supernatant is withdrawn and checked by UPLC or GC-MS to confirm reaction completion (Note 27). The flask containing the red biphasic mixture (Figure 3D) is removed from the oil bath and allowed to cool to 22 ℃. The stir bar is retrieved using a magnetic stir bar retriever. The reaction mixture is then transferred to a 250-mL separatory funnel and the flask is rinsed with ethyl acetate (3 x 10 mL) followed by deionized water (10 mL). The aqueous and organic phases are shaken, separated and the aqueous phase is extracted twice more with 20 mL portions of ethyl acetate. The organic phases (totaling 80 mL) are combined in a 250-mL Erlenmeyer flask and dried over anhydrous sodium sulfate (15 g), before filtration through a 150-mL filter funnel (glass frit, medium porosity) into a 250-mL round-bottomed flask. The contents of the funnel are washed with ethyl acetate (3 x 10 mL). The filtrate is removed of volatiles via rotary evaporation under reduced pressure (45 ℃ water bath, 90 mmHg).
Using a glass column (6 cm in diameter x 18 cm in height), a silica gel column is prepared using 140 g of silica gel and primed with 500 mL of hexanes. The crude product is dissolved in 10 mL of methylene chloride and applied to the column. The flask containing the residual crude product is rinsed twice with 5 mL portions of methylene chloride, which are loaded to the column. A 4 cm layer of sand is placed on top of the silica gel (Figure 4A). The column is initially eluted with 500 mL of hexanes, followed by eluting with a 9:1 mixture of hexanes:ethyl acetate. The fractions containing the product (a total of 950 mL) (Note 28) are collected and concentrated by rotary evaporation under reduced pressure (using a 40 ℃ water bath, from 320 mmHg to 70 mmHg). The resulting white crystalline product is further dried under high vacuum (<10 mmHg) for 16 h, yielding 4-acetylbiphenyl (6) (2.41 g, 98%) (Figure 4B) (Notes 29 and 30).

Figure 4. A. Column loaded with crude material; B. Isolated 4-acetylbiphenyl product (6). (Photos provided by the submitting authors)
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/about/governance/committees/chemical-safety.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
diphenylphosphine (
1),
3-chloro-1-propanol (
2),
n-butyllithium solution (2.5 M in hexanes),
4'-bromoacetophenone (
4),
phenylboronic acid (
5),
bis(1,5-cyclooctadiene)nickel (
Ni(cod)2),
potassium phosphate tribasic (
K3PO4), and
2-methyltetrahydrofuran (
2-MeTHF).
2.
3-Chloro-1-propanol (
2, >98%) was purchased from Oakwood Chemical and used as received.
3.
THF (anhydrous, HPLC grade, purchased from Caledon Laboratories Ltd.) was deoxygenated and dried by using an Inert Technologies solvent purification system.
4.
Diphenylphosphine (
1, 98%) was purchased from Sigma-Aldrich and used as received. This compound is sensitive to air and undergoes oxidation to P(O)Ph
2H when exposed outside of the glovebox. Therefore, storing it in the glovebox is recommended.
5.
n-Butyllithium solution (
n-BuLi, 2.5 M in hexanes) was purchased from Sigma-Aldrich and used as received.
6. When
n-BuLi was added to the reaction, an immediate color change to an orange color occurred, but the color quickly disappeared. The color change was attributed to the initial reaction of
n-BuLi with
PPh2H, forming orange LiPPh
2 in
THF.
7. The reaction progress was monitored using TLC, using a 4:1 mixture of hexanes:
ethyl acetate as the eluent. The plate was visualized under a 254 nm UV lamp. The starting material,
PPh2H (
1), has an R
f value of 0.85, while the
ProPhos product (
3) shows an R
f value of 0.30 (Figure 5).
Figure 5. TLC of the crude reaction mixture: SM = starting material 3-chloropropanol (2), CO = co-spot of SM and RX, RX = reaction mixture. (Photos provided by the submitting authors)
8. Deoxygenated
NH4Cl (aq.) was prepared by sparging a saturated aqueous
NH4Cl solution in a Schlenk flask with nitrogen gas for 30 min.
NH4Cl was purchased from Sigma-Aldrich and used as received.
9.
Ethyl acetate was purchased from Sigma-Aldrich and used as received.
10. Anhydrous
sodium sulfate (
Na2SO4) was purchased from Sigma-Aldrich and used as received.
11. Silica gel (SiliaFlash F60) was purchased from SiliCycle, Inc. and used as received.
12. Hexanes was purchased from Fisher Chemical and used as received.
13.
Methylene chloride was purchased from Sigma-Aldrich and used as received.
14. TLC of the fractions used a 4:1 hexanes:
ethyl acetate solvent system as eluent (Figure 6). Fractions 32-56 (each fraction ~25 mL) containing the target compound were pooled in a 1 L round-bottom flask. To ensure complete transfer, each fraction test tube was rinsed twice with 1 mL of
methylene chloride.
Figure 6. Fractions collected. (Photos provided by the submitting authors)
15. The checking authors found that the temperature of the water bath must be carefully controlled. Prolonged heating of the purified material in air should be avoided, as it accelerates the oxidation of
ProPhos and decreases the purity.
16. Precise yields for each run were 3.47 g (88%) and 3.71 (94%), respectively. The purity of product
3 obtained from chromatography for both runs was determined to be 97.0% by qNMR
pdf using
1,3,5-trimethoxybenzene (99.8%) (
Note 18) as the internal standard.
17. The product
3 is characterized as follows: mp. 59 - 60 ℃;
1H NMR
pdf (400 MHz, CDCl
3) δ 7.46 - 7.42 (m, 4H), 7.36 - 7.30 (m, 6H), 3.70 (t,
J = 6.4 Hz, 2H), 2.13 (m, 2H), 1.70 (ddt,
J = 15.7, 9.3, 6.4 Hz, 2H) ppm;
31P{
1H} NMR
pdf (162 MHz, CDCl
3) δ -16.2 (s) ppm;
13C{
1H} NMR
pdf (101 MHz, CDCl
3) δ 138.5 (d,
J = 12 Hz), 132.8 (d,
J = 19 Hz), 128.9 (s), 128.5 (d,
J = 7 Hz), 63.6 (d,
J = 14 Hz), 29.1 (d,
J = 15 Hz), 24.3 (d,
J = 11 Hz) ppm; IR (ATR, neat): 3381, 3069, 2934, 2896, 2871, 1480, 1431, 1303, 1195, 1173, 1009, 828, 733 cm
-1; HRMS (DART, CH
3CN) m/z: [M + H]
+ calcd for C
15H
18OP: 245.1090, found: 245.1089.
18.
1,3,5-Trimethoxybenzene (99.8%) was purchased from Sigma-Aldrich (standard for quantitative NMR, TraceCERT®, manufactured by: Sigma-Aldrich Production GmbH, Switzerland) and used as received.
19.
Bis(1,5-cyclooctadiene)nickel(0) (
Ni(cod)2) was purchased from Strem Chemicals and used as received. This compound is extremely air sensitive and the generation of Ni precatalyst must be conducted in the glovebox.
20. The submitting authors originally used 0.1 mol%
Ni(cod)2 catalyst and 0.4 mol%
ProPhos (
3), however the checking authors found that the reaction was more reproducible at a slightly higher catalyst loading, due to the extreme air sensitivity of the
Ni(cod)2 precatalyst.
21.
2-Methyltetrahydrofuran (>99%, extra dry over molecular sieves, stabilized, AcroSeal
TM) was purchased from Thermo Scientific Chemicals and used as received.
22.
4'-Bromoacetophenone (
4, 99%) was purchased from Oakwood Chemical by the checking authors and used as received. The submitting authors purchased aryl bromide
4 from Ambeed (99%).
23.
Phenylboronic acid (
5, 95%) was purchased from Sigma-Aldrich and used as received.
24.
Potassium phosphate tribasic (
K3PO4) (>98%) was purchased from Sigma-Aldrich and used as received.
25. The checking authors found that addition of the Ni precatalyst to the reaction mixture within the glovebox could mitigate potential oxygen contamination during the addition process. Alternatively, the submitting authors withdrew the Ni precatalyst using a plastic syringe. The needle tip was inserted into a rubber septum for protection, then the filled syringe assembly was taken out of the glovebox and quickly injected to the reaction mixture outside the glovebox.
26. Deionized water was sparged vigorously with nitrogen gas for at least 30 min before use.
27. The reaction progress is difficult to track using TLC analysis on silica gel, with the starting material
4'-bromoacetophenone (
4) and the product (
6) having a similar R
f value of 0.45 using a 9:1 hexanes:
ethyl acetate eluent, visualized using a 254 nm UV lamp. (Figure 7). The submitting authors found that GC-MS analysis of the reaction mixture shows the complete consumption of the starting material
4'-bromoacetophenone. Alternatively, the checking authors found that UPLC analysis of the reaction mixture also shows complete starting material consumption.
Figure 7. TLC of the crude reaction mixture (SM = starting material 4, CO = co-spot of SM and RX, RX = reaction mixture) (Photo provided by the submitting authors)
28. Fractions containing the desired product
6 were identified using TLC eluting with a 9:1 hexanes:
ethyl acetate solvent system (Figure 8). Fractions 22-40 (~50 mL each test tube), which contained the target compound, were pooled into a 1 L round-bottom flask. To ensure complete transfer, each fraction was rinsed twice with 1 mL of
methylene chloride.
Figure 8. Fractions collected. (Photos provided by the submitting authors)
29. Precise yields for each run were 2.33 g (94%) and 2.41 g (98%). The purity of product
6 obtained from chromatography was determined to be 99.3 and 100.0 wt% respectively by qNMR
pdf using
1,3,5-trimethoxybenzene (99.8%) (
Note 18) as the internal standard.
30. Product
6 is characterized as follows: mp. 120 - 121 ℃;
1H NMR
pdf (400 MHz, CDCl
3) δ 8.04 (d,
J = 8.9 Hz, 2H), 7.79 (d,
J = 8.1 Hz, 2H), 7.63 (d,
J = 7.6 Hz, 2H), 7.48 (t,
J = 7.4 Hz, 2H), 7.41 (t,
J = 7.4 Hz, 1H), 2.64 (s, 3H) ppm;
13C{
1H} NMR
pdf (101 MHz, CDCl
3) δ 197.8 (s), 145.8 (s), 139.9 (s), 135.9 (s), 129.1 (s), 129.0 (s), 128.3 (s), 127.4 (s), 127.3 (s), 26.7 (s) ppm; IR (ATR, neat): 2998, 2918, 1677, 1600, 1402, 1358, 1282, 1207, 1178, 959, 832, 763 cm
-1; HRMS (DART, CH
3CN) m/z: (DART, CH
3CN) m/z: [M + H]
+ calcd for C
14H
13O: 197.0961, found: 197.0955.
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
This procedure outlines the synthesis of a ligand, ProPhos, that enables an efficient and scalable nickel-catalyzed Suzuki-Miyaura coupling (Ni-SMC) of aryl halides and boronic acids or esters, in green solvents 2-methyltetrahydrofuran (
2-MeTHF)/water mixture or 2-propanol (
i-PrOH). The Suzuki-Miyaura coupling is extensively applied in pharmaceutical syntheses of small molecule drugs.
3 While palladium catalysts are currently used in process syntheses, Ni-SMC has garnered significant attention due to its sustainability and low cost.
4,5,6,7 However, the application of Ni-SMC has been limited by a slow reaction rate,
8 high catalyst loadings,
4,6 the need for expensive, air-sensitive precatalysts, and a limited scope for heterocycles.
9,10 Furthermore, achieving cross-coupling reactions in environmentally benign solvents continues to be a critical objective in green chemistry.
11,12 It is noteworthy that organic solvents account for up to 85% of the waste generated during drug synthesis.
13We have developed a cost-effective, efficient, and user-friendly phosphine ligand, 3-(diphenylphosphino)propanol (ProPhos) (
3), to address these challenges in Ni-SMC (Figure 13).
14 A preliminary analysis estimates the cost for ProPhos to be $1.50/g based on this procedure. Notably, the cost of ProPhos could be further reduced to $0.34/g if it is synthesized from triphenylphosphine and lithium metal, although the procedure would be less user-friendly.
Transmetalation is considered to be the turnover-limiting step in Ni-SMC.
14,15,16 ProPhos
3 promotes transmetalation by its pendent hydroxyl group which coordinates with boronic acids or esters, thereby promoting colocation and preorganization of the nickel catalyst and the boronic acid or ester for efficient transmetalation (Figure 9). Alternatively, transmetalation can be promoted through intramolecular ligand substitution, where the halide is displaced to form a Ni-oxo species, leading to fast transmetalation via a Ni-O-B interaction. Furthermore, the hydroxyl group of ProPhos may act as a hemilabile ligand, offering protection against catalyst deactivation through coordination with heteroaryl substrates and products,
17 while still readily dissociating to preserve high catalytic efficiency.
Figure 9. Functions of the ProPhos ligand in Ni-SMC
ProPhos can accommodate both Ni(cod)
2 and the inexpensive, air-stable nickel(II) chloride hexahydrate (NiCl
2·6H
2O) ($0.06/g)
18,19,20 as precatalysts (Figure 10). These (ProPhos)Ni catalysts demonstrate rapid kinetics and robust activity across reactions with various heteroarenes, including pyridine, quinoline, pyrazole, pyrimidine, indole and 2-aminopyridine, with catalyst loadings ranging from 0.1 to 3 mol %. It is worth noting that several previous studies have reported Ni-SMC utilizing low nickel loadings (≤1 mol %).
19,20,21,22,23,24 The reactions were also carried out in green solvents, either 2-MeTHF or
i-PrOH. A mixed solvent system of 2-MeTHF/H
2O with Ni(cod)
2 and ProPhos exhibits good activity with arylboronic acid pinacol esters (ArBPin) and non-heteroaryl boronic acids. For heteroaryl boronic acids, the catalytic reactions proceed well with NiCl
2·6H
2O and ProPhos in
i-PrOH.
19,20 In pharmaceutical process synthesis, substituting Ni(cod)
2 with an air-stable and cost-effective nickel precursor, such as NiCl
2·6H
2O, is highly desirable for large-scale applications.
4
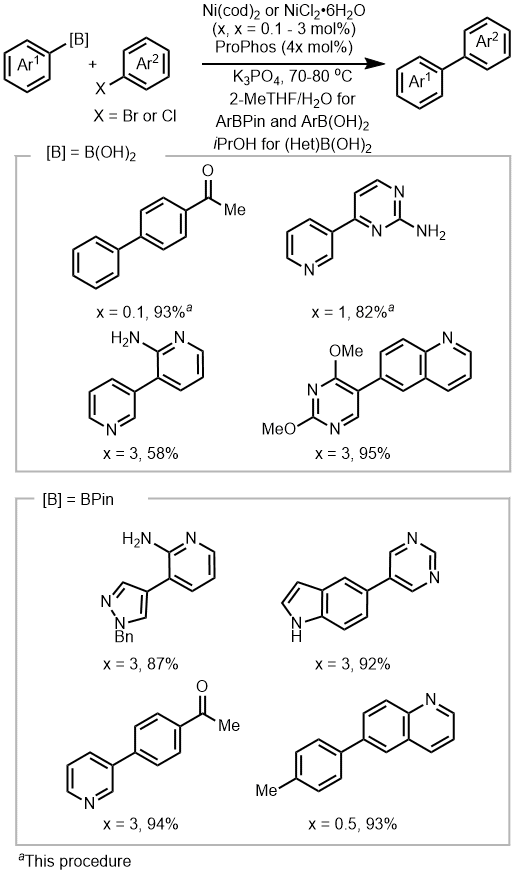
Figure 10. Selected scope of (ProPhos)Ni catalyzed SMC
14In summary, this protocol describes the synthesis of a phosphine ligand, ProPhos, which facilitates Ni-SMC in green solvents. (ProPhos)Ni offers potential for applications in Ni-SMC in pharmaceutical process synthesis.
Appendix
Chemical Abstracts Nomenclature (Registry Number)
Diphenylphosphine (1) (829-85-6)
3-Chloro-1-propanol (2) (627-30-5)
n-Butyllithium solution (2.5 M in hexanes) (109-72-8)
3-(Diphenylphosphino)-1-propanol (3) (2360-09-0)
4'-Bromoacetophenone (4) (99-90-1)
Phenylboronic acid (5) (98-80-6)
Bis(1,5-cyclooctadiene)nickel (1295-35-8)

|
Jin Yang graduated with a first-class honors B.Sc. in Chemistry from St. Francis Xavier University in Canada. He then pursued his M.Sc. and Ph.D. at the University of Victoria, Canada, under the guidance of Professor Lisa Rosenberg, focusing on organometallic and inorganic chemistry. In 2023-2025, he joined New York University as a postdoctoral fellow, working with Professor Tianning Diao on metal catalysis and organic reaction discovery. He is now an assistant professor at Yeshiva University. |

|
Rui Zhang was born and raised in Xingtai, Hebei Province, China. He earned his B.Sc. from Fudan University in 2017. He then pursued a Ph.D. at the Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, under the supervision of Professor Kuiling Ding, focusing on Pd-catalyzed asymmetric catalysis. In 2024, he moved to the United States to join Professor Tianning Diao's group at New York University as a postdoctoral researcher, where he is currently working on solving challenging problems in organic reactions. |

|
Tianning Diao was born and raised in Chengdu, Sichuan Province, China. She earned her B.Sc. from Fudan University in 2007 and completed her Ph.D. at the University of Wisconsin-Madison, under the supervision of Professor Shannon Stahl. After postdoctoral training with Paul Chirik at Princeton University, she started her independent career at New York University in 2014, where she is currently a Professor of Chemistry. Her research focuses on developing nickel-catalyzed cross-coupling reactions and exploring their reaction mechanisms. |

|
Benjamin Chiu received his B.Sc. in Chemistry from the University of Toronto. He began his Ph.D. with Professor Sophie Rousseaux at the University of Toronto in 2022. His current research is focused on developing Ni-catalyzed cyanation reactions for the synthesis of nitrile compounds. |

|
Sophie Rousseaux is an Associate Professor of Chemistry at the University of Toronto, where she also holds a Canada Research Chair (Tier 2) in Organic Chemistry and serves on the Scientific Leadership Team of the Acceleration Consortium. Her group's research interests include organic synthesis, catalysis, and organometallic chemistry, with a particular focus on the synthesis of small rings and nitrile-containing molecules. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved