Org. Synth. 2026, 103, 253-268
DOI: 10.15227/orgsyn.103.0253
Preparation of Enantiopure Bifunctional S(VI) Transfer Reagent (t-BuSF)
Submitting Authors: Zachary Shultz, Eve Hall, Prakash Warghude, Yun-Pu Chang, Justin M. Lopchuk*
1Checking Authors: Taylor A. Stephens and Tehshik P. Yoon*
21. Procedure (Note 1)
A. N,N-Bis(1-methylethyl)carbamic chloride (2). (Note 2). A 250-mL, three-necked, round-bottomed flask equipped with a 3 x 1.5-cm Teflon-coated magnetic stir bar, 125-mL pressure-equalizing addition funnel fitted with a rubber septum, ground-glass fitted adapter containing a thermometer, and a rubber septum under a positive pressure of nitrogen is charged with anhydrous dichloromethane (DCM, 30 mL) (Note 3), and cooled to 0 ℃ with an ice-water bath. Bis(trichloromethyl) carbonate (triphosgene, 1) (6.02 g, 20.3 mmol, 1 equiv) (Note 4) is added in one portion (Note 5), and the addition funnel was charged with a previously prepared solution of anhydrous i-Pr2NH (8.6 mL, 61 mmol, 3 equiv) (Note 6) and anhydrous Et3N (8.6 mL, 61 mmol, 3 equiv) (Note 7) in DCM (90 mL) under an atmosphere of nitrogen (Note 8). At this time, magnetic stirring (450 rpm) is started (Note 9) along with slow addition over 10-15 min of the amine solution to maintain an internal temperature below 25 ℃ (Note 10) to give a pale-yellow suspension. Vigorous stirring continues for 1 h at 0 ℃.

Figure 1. A. Reaction apparatus; B. Addition of bis(trichloromethyl) carbonate (1); C. Addition of the amine solution; D. Color change and solid formation during addition of the amine solution.
After this time, the reaction mixture is removed from the ice bath, the addition funnel is removed and the solids (Note 11) filtered via vacuum filtration through a medium-porosity sintered-glass 150-mL funnel. The round-bottomed flask and the filter-cake are rinsed with DCM (2 x 20 mL), and the filtrate is collected into a 500-mL round-bottomed flask and concentrated by rotary evaporation (25 ℃) (Note 12) to give a yellow oily suspension. Hexanes (250 mL) (Note 13) is added to dissolve the crude mixture that is then transferred to a 500-mL separatory funnel and washed with water (3 x 60 mL). The hexanes layer is transferred to a 500-mL Erlenmeyer flask where it is dried over anhydrous sodium sulfate (20 g) and filtered, using a plastic funnel fitted with a cotton plug, into a pre-weighed 500 mL round-bottomed flask. Solvents are removed by rotary evaporation (300 mbar, 25 ℃) to obtain the desired product 2 as a white crystalline solid (6.54 g, 40.0 mmol, 65% yield) (Note 14).

Figure 2: A. Vacuum filtration of solids; B. Suspension formed during rotary evaporation of DCM. C. Hexanes and water layers during work-up; D. Collected organic layer dried over Na2SO4; E. Solid product obtained after removal of hexanes; F. Storage of product
B. (S)-N-(Diisopropylcarbamoyl)-2-methylpropane-2-sulfonimidoyl fluoride, t-BuSF (4) (Note 15). A 500-mL, three-necked, round-bottomed flask equipped with a 3 x 1.5-cm Teflon-coated magnetic stir bar, 125-mL pressure-equalizing addition funnel fitted with a rubber septum, ground-glass fitted adapter containing a thermometer, and a rubber septum under a positive pressure of nitrogen is charged with NaH (4.01 g, 100 mmol, 2.52 equiv, 60% wt;) (Note 16) and cooled to 0 ℃ in an ice-water bath. Anhydrous THF (100 mL) (Note 17) is added to the addition funnel and carefully dispensed (Note 18) to give a gray suspension. A solution of (R)-2-methylpropane-2-sulfinamide 3 (5.06 g, 41.7 mmol, 1.05 equiv) (Note 19) in THF (100 mL) was added to the addition funnel (Note 20) and dispensed slowly over 10-15 min (Note 21) to give a light-grey suspension. The flask with the solution of 3 in THF is rinsed with THF (10 mL) and added to the addition funnel. It was then added slowly over 2 min to the reaction mixture and stirred at 0 ℃ for 30 min.

Figure 3: A. Reaction set-up; B. Charged with NaH and THF; C. Solution of compound 3 in THF; D. Suspension of NaH in THF and charged addition funnel with solution of compound 3; E. Addition of compound 3 to NaH suspension; F. Rinsed additional funnel with THF after adding compound 3
While cooling in an ice-water bath, solid N,N-bis(1-methylethyl)carbamic chloride (2) (7.17 g, 43.8 mmol, 1.1 equiv) is added in two portions (3.58 g x 2) to give a turbid beige solution (Note 22) and stirred for 1 h at the same temperature (Note 23). The reaction progress is monitored by TLC (Note 24) using 3 as a standard, which is consumed within 1 h. At this time, solid N-fluorobenzenesulfonimide (NFSI) (12.52 g, 39.7 mmol, 1 equiv) (Note 25) is added in one portion (Note 26) to give a white suspension that stirs for 30 min in an ice-water bath. The reaction progress is monitored by TLC (Note 27).
Figure 4: A. Addition of compound 2 as a solid; B. The resulting reaction mixture and generation of H2 gas after the addition of 2; C. TLC of the reaction mixture 1 h after the addition of compound 2; D. Addition of NFSI as a solid; E. The resulting white suspension after the addition of NFSI; F. TLC of the reaction mixture 30 min after the addition of NFSI
A solution of EtOAc/hexanes (20% EtOAc in hexanes, 150 mL) (Note 28) is added and stirring continues for 5 min at 0 ℃ to facilitate transfer and filtration of by-products (Note 29). The resulting white suspension is vacuum filtered (house vacuum) through a 500-mL medium-porosity sintered glass funnel containing a pad of Celite (3-cm thick). The three-neck round-bottomed flask and filter-cake is rinsed with a 20% EtOAc/hexanes (2 x 50 mL). The filtrate is transferred to a 1-L separatory funnel, washed with saturated aqueous NH4Cl (100 mL) and saturated aqueous Na2S2O3 (2 x 100 mL) (Note 30). The combined aqueous layers are extracted with hexanes/EtOAc (20% EtOAc/hexanes, 3 x 75 mL) and combined with the first organic layer. The combined organic layers are dried over Na2SO4 (25 g), filtered through a cotton-plug in a plastic funnel, and the filtrate is concentrated by rotary evaporation (300 mbar, 25 ℃) to give a golden oil.

Figure 5: A. The reaction mixture after dilution with 20% EtOAc in hexanes; B. Sinter-glass funnel with Celite filter ; C. Solids removed by filtration; D. Reaction mixture partitioned in a 1-L separatory funnel with saturated aqueous NH4Cl; E. Organic and aqueous layers after work-up; F. Crude oil obtained after work-up and removal of solvents; G. TLC analysis of four racks of test-tubes collected during column chromatography. H. Compound 4 after purification
The crude oil is purified by flash-column chromatography with hexanes/EtOAc (0% to 20% EtOAc) as the solvent system (Notes 31 and 32). Compound 4 is obtained as a clear oil that solidifies to a white crystalline solid upon standing (manual column chromatography provided 7.80 g, 29.0 mmol, 74% yield) (Notes 33 and 34).
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regards to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/about/governance/committees/chemical-safety.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
bis(trichloromethyl) carbonate (see
Note 4),
dichloromethane,
triethylamine,
N,N-diisopropyl amine, hexanes,
sodium sulfate,
tetrahydrofuran,
sodium hydride,
tert-butyl sulfinamide,
N-fluorobenzenesulfonimide,
Celite,
ethyl acetate,
ammonium chloride,
sodium thiosulfate, silica gel.
2. All the glassware used for this preparation was dried in an oven at 200 ℃ for at least two hours prior to use.
3. Anhydrous
dichloromethane (
DCM) was purchased from Fisher Scientific (HPLC grade, 99.9% purity) and obtained by passing the solvent through an activated alumina column (PPT Glass Contour Solvent Purification System).
4.
Bis(trichloromethyl) carbonate (>99% purity), "triphosgene" or "BTC", was purchased from Chem-Impex and used as received. The solid reagent triphosgene is a less hazardous substitute for highly toxic gaseous phosgene. Extreme care should be used when handling triphosgene; may be fatal if inhaled and causes burns by all exposure routes. Upon exposure to water, triphosgene liberates toxic gases phosgene and HCl. Triphosgene is a lachrymator and decomposes violently at elevated temperatures. Triphosgene should be weighed in a well-ventilated area and the reaction should be performed in a well-ventilated fume hood. See
Organic Process Research & Development 2017 21 (9), 1439-1446. DOI: 10.1021/acs.oprd.7b00220 for further discussion.
5.
Bis(trichloromethyl) carbonate (
1) is added to pre-cooled
DCM at 0 ℃ without stirring.
6.
Diisopropylamine (
i-Pr2NH, >99% purity) was purchased from Oakwood Chemicals and obtained by passing the solvent through an activated alumina column (PPT Glass Contour Solvent Purification System).
7.
Triethylamine (
Et3N, 99.5% purity) was purchased from Oakwood Chemicals and obtained by passing the solvent through an activated alumina column (PPT Glass Contour Solvent Purification System).
8. A stock solution of the amine and solvent was prepared in a flame dried 250 mL round bottom flask by syringe transfer under a positive atmosphere of nitrogen. The required amount was then transferred by 50 mL syringe with an 18-gauge metal needle to the addition funnel.
9.
Bis(trichloromethyl) carbonate (
1) dissolved in
DCM upon stirring.
10. Gas evolution occurs upon addition of the amine solution with an observable exotherm temporarily raising the internal temperature to 23 ℃. The temperature of the reaction mixture returned to 0 ℃ once the addition was complete.
11. The solids removed are the hydrochloride salts of
Et3N and unreacted
i-Pr2NH.
12. The round-bottomed flask was removed from the rotary evaporator once
DCM stopped condensing. Rigorous evaporation of the unreacted amine is not required. The distillate may contain residual phosgene and caution should be used with handling and disposal. Any remaining phosgene is quenched during aqueous work-up. Sodium bicarbonate is added to the collection flask during evaporation as a safety precaution.
13. Hexanes (ACS grade, 99.9% purity) was purchased from Fisher Scientific and used as received.
14. Analytical data for
N,N-bis(1-methylethyl)carbamic chloride (
2):
1H NMR
pdf (500 MHz, CDCl
3) δ 4.55 (hept, J = 6.6 Hz, 1H), 3.59 (hept, J = 6.2 Hz, 2=1H), 1.37 (d, J = 6.7 Hz, 6H), 1.22 (d, J = 6.7 Hz, 6H);
13C NMR
pdf (126 MHz, CDCl
3) δ 146.0, 52.9, 48.6, 20.4, 20.0; mp: 57-58 ℃. Purity for the two runs were determined to be 98 and 100% respectively by qNMR
pdf using
1,3,5-trimethoxybenzene (99%, TCI Chemicals) as an internal standard.
15. All the glassware used for this preparation was dried in an oven at 200 ℃ for at least two hours prior to use. The set up was evacuated and backfilled with nitrogen twice for about 3 min.
16.
Sodium hydride (
NaH) was purchased from Thermo Fisher Scientific (57-63% wt, oil dispersion), stored under argon, and used as received. A weight percent of 60% was to calculate the mass for 2.5 equiv. Excess (0.5 equiv) of
NaH is used to assure full deprotonation of sulfinamide
3 and the sulfinyl urea intermediate that forms during the course of the reaction.
17. Anhydrous
tetrahydrofuran (
THF) was purchased from Fisher Scientific (HPLC grade, 99.9% purity) and obtained by passing the solvent through an activated alumina column (PPT Glass Contour Solvent Purification System).
18.
THF was transferred by 50 mL syringe with an 18-gauge metal needle.
19.
(R)-2-Methylpropane-2-sulfinamide (
3) was purchased from Ambeed (98% purity, >99%
e.e.).
20.
THF was transferred using a 50 mL syringe with an 18-gauge metal needle to a flame dried 500 mL round bottom flask under positive nitrogen pressure containing
(R)-2-methylpropane-2-sulfinamide (
3). The solution was then transferred to the addition funnel by syringe.
21. The evolution of
H2 gas is observed immediately along with an exotherm temporarily raising the internal temperature to 7-8 ℃.
22. The evolution of
H2 gas is observed approximately 3 min after the addition of
2 along with an exotherm temporarily raising the internal temperature to 8 ℃. The reaction mixture changes to turbid beige solution once
H2 gas evolution ceases.
23. Removal of excess water and additional ice is added to the ice-water bath to maintain cooling at 0 ℃.
24. The reaction is sampled directly for TLC analysis using 30% acetone in hexanes as the solvent system with
(R)-2-methylpropane-2-sulfinamide 3 as a standard. Ninhydrin stain was used to visualize the target intermediate. The
tert-butyl sulfinyl urea intermediate is not stable to aqueous work-up conditions.
25.
N-Fluorobenzenesulfonimide (
NFSI) was purchased from Ambeed (98 % purity). We have observed a small impurity by
19F NMR (50.5 ppm, up to 2% relative integration) in the final product depending on the source of
NFSI. No notable decrease in the quality or efficiency of
t-BuSF has been observed if this contaminant is present.
26. An exotherm is observed raising the internal temperature temporarily to 24-28 ℃ depending on the rate at which
NFSI is added. Faster addition results in a larger exotherm. No negative impacts to yield or purity have been observed with varying rates of addition.
27. An aliquot (20 μL) of the reaction mixture was taken and partitioned in
EtOAc/saturated aqueous
NH4Cl (200/500 μL) for TLC analysis using 20%
EtOAc in hexanes as the solvent system with
NFSI and
tert-butyl sulfinamide 3 as standards. The TLC plate was observed under long-wave UV light to monitor
NFSI consumption. Ninhydrin stain was used to visualize the target product and other impurities.
28. Hexanes (ACS grade, 99.9% purity) and
ethyl acetate (
EtOAc, ACS grade, 99.9% purity) was purchased from Fisher Scientific and used as received.
29. Sodium
N-benzenesulfonimidate is the solid by-product. Caution: the filtered solids may contain unreacted
NaH. The filter cake should be quenched with 5 mL of methanol before evaporation of residual solvent and prior to disposal.
30.
Sodium thiosulfate (
Na2S2O3) is used to quench any remaining
NFSI present in the reaction mixture.
31. Manual flash-column chromatography was performed for two trials using the following parameters by the checking authors: glass column (35/20 mm) packed with 100 g of silica gel (40-63 μm particle size) wet with hexanes; crude oil loaded directly and the round-bottomed rinsed with hexanes (2 x 10 mL). Sand (20 g) is added on top of the silica gel followed by gradient elution: hexanes (300 mL), 5%
EtOAc in hexanes (100 mL), 10%
EtOAc in hexanes (250 mL), 15%
EtOAc in hexanes (150 mL), 20%
EtOAc in hexanes (700 mL), with a flowrate of 20-30 mL/min. The desired product began eluting with 15%
EtOAc in hexanes. Column fractions of size 16 x 150 mm were used. Fractions 30 - 45 had product.
32. Automated flash-column chromatography was performed for two trials on a Biotage Selekt system using the following parameters by the submitting authors: Biotage Sfär 100 g plastic column (50 mm i.d.) packed with 100 g of silica gel (40 -63 mm particle size) topped with a frit. Column equilibration with hexanes; crude oil loaded directly and the round-bottomed rinsed with hexanes (2 x 10 mL). A solvent gradient of 0% to 13%
EtOAc (5 column volumes) holding at 13%
EtOAc for 5 column volumes then to 20% (3 column volumes), with a flowrate of 120 mL/min. The desired product began eluting at 12%
EtOAc in hexanes. First and second runs using automated column chromatography: 9.05 g, 34.0 mmol, 86% yield; 9.10 g, 34.2 mmol, 86% yield. Purity for the two runs were determined to be 98% and 99% respectively by qNMR using
1,3,5-trimethoxybenzene (99%, TCI Chemicals) as an internal standard.
33. Analytical data for
N-(diisopropylcarbamoyl)-2-methylpropane-2-sulfonimidoyl fluoride,
t-BuSF (
4):
1H NMR
pdf (500 MHz, CDCl
3) δ 4.10 - 3.98 (m, 1H), 3.94 - 3.82 (m, 1H), 1.60 (s, 9H), 1.29 (d, J = 5.9 Hz, 6H), 1.22 (d, J = 6.0 Hz, 6H).
13C NMR
pdf (126 MHz, CDCl
3) δ 153.8, 62.7, 62.6, 47.7, 45.8, 21.2, 24.6, 20.6, 20.5.
19F NMR
pdf (377 MHz, CDCl
3) δ 33.2.; mp: 72-74 ℃. Purity for the two runs were determined to be 100% and 100% respectively by qNMR
pdf using
1,3,5-trimethoxybenzene (99%, TCI Chemicals) as an internal standard. specific rotation [α]
20/
D = +89.50 (c 6.93, CH
2Cl
2); HRMS: Calc'd for C
11H
23FN
2NaO
2S [M+Na
+] 289.1357; found 289.1353. Enantiomeric excess determined to be >99%
e.e. by chiral HPLC
pdf (Daicel Chiralpak IC column, 70:30
n-hexane:
i-PrOH, flow rate: 1 mL/min, 25 ℃, UV detection wavelength: 210 nm, retention time: minor: 12.9 min, major: 16.3 min. Racemic material made using the same procedure but with racemic 2-methyl-2-propanesulfinamide.
34. Yields on half scale: 3.13 g, 19.1 mmol, 60% yield (Step 1); 3.14 g, 17.2 mmol, 83% yield (Step 2).
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
The discovery of
t-BuSF (
4) provided a crystalline, bench-stable, enantiopure bifunctional S(VI) transfer reagent that could overcome long-standing challenges in accessing sulfoximines, sulfonimidoyl fluorides, and sulfonimidamides with stereocontrol.
3 Enabled by a novel urea-based sulfonimidoyl protecting group and prepared in a single step from
tert-butyl sulfinamide (
3),
t-BuSF (
4) (>99%
e.e.) undergoes asymmetric SuFEx with organolithiums to
tert-butyl sulfoximines (
5). These stereogenically stable sulfoximines serve as activatable synthons to strategically access S(IV) and S(VI) derivatives through stereospecific reactions with carbon and nitrogen nucleophiles. In the initial disclosure, the scope of and utility of
t-BuSF (
4) was demonstrated in over 70 examples
, including sulfoximine and sulfonimidamide analogs of celecoxib and begacestat, highlighting both synthetic efficiency and translational relevance.
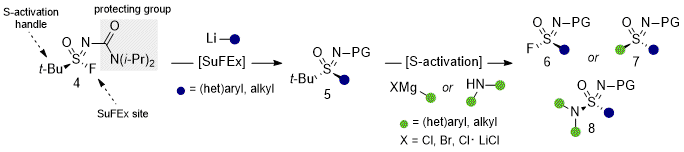
Figure 6: Design and use of t-BuSF (4) for the asymmetric synthesis of sulfonimidoyl functionality. SuFEx conditions: Et2O, -78 or -40 ℃; S-activation conditions: t-BuOK, THF, 80 ℃ then AcOH, NFSI, THF 0 ℃ or TFA, DCM, rt then t-BuOK, NFSI, THF, 0 ℃
During the developmental of
t-BuSF (
4), urea-based protecting groups were found to increase the stability and SuFEx reactivity for carbon and nitrogen nucleophiles, with the
N,
N-diisopropyl derivative proving superior. In a subsequent study, the diisopropyl urea protecting group was leveraged as a third point of diversification.
4 Through a strain-release driven process, the
N,
N-diisopropyl amine was efficiently exchanged with primary and secondary amines of sulfinyl and secondary sulfonimidamide urea intermediates-enabling stereospecific access to functionalized sulfinamides, sulfonimidamides, and sulfoximines in high enantiopurity (up to >99%
e.e.). This strategy was validated across more than 50 examples with improved accessibility to pharmaceutically relevant sulfonimidoyl ureas affording reduced step-counts and the addition of a third modular site for structural diversification. Additionally, the multi-functionalization sequences were amenable to combinatorial workflows, further underscoring the breadth of stereodefined S(VI) chemistry achievable from a single reagent platform.
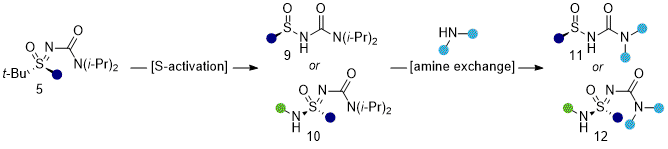
Figure 7: Accessing sulfinyl and sulfonimidoyl urea intermediates from tert-butyl sulfoximines and their functionalization via N,N-diisopropyl amine exchange
Taken together, these advances illustrate two complementary design principles unified by t-BuSF (4): bench stability and modularity, which facilitate step-economic asymmetric synthesis, and designed strain-release reactivity, which drives transformations not otherwise accessible. This duality positions t-BuSF (4) as a uniquely versatile reagent, expanding the synthetic and medicinal chemistry potential of stereocontrolled S(IV) and S(VI) scaffolds.
Appendix
Chemical Abstracts Nomenclature (Registry Number)
bis(trichloromethyl) carbonate (1) (32315-10-9)
Triethylamine (121-44-8)
Diisopropylamine (108-18-9)
N,N-Bis(1-methylethyl)carbamic chloride (2) (19009-39-3)
(R)-2-Methylpropane-2-sulfinamide; (3) (196929-78-9)
Sodium hydride (7646-69-7)
N-Fluorobenzenesulfonimide (133745-75-2)
(S)-N-(diisopropylcarbamoyl)-2-methylpropane-2-sulfonimidoyl fluoride; t-BuSF: 2-Propanesulfonimidoyl fluoride, N-[[bis(1-methylethyl)amino]carbonyl]-2-methyl-, [S(S)]- (ACI) (4) (2935590-83-1)

|
Justin M. Lopchuk obtained his PhD in 2013 from Dartmouth College in the group of Prof. Gordon W. Gribble. He then completed his postdoctoral fellowship in the group of Prof. Phil S. Baran at The Scripps Research Institute. In 2017, he began his independent career at the H. Lee Moffitt Cancer Center & Research Institute where he is now an Associate Professor focusing on the development of new methods for C-C, C-N, and C-S bond formation, novel chemistry for covalent inhibition, natural product synthesis, and structure-based drug design. |

|
Zachary Shultz obtained his PhD in 2019 from the University of South Florida (USA), in the group of Professor James Leahy, with a research focus on natural product total synthesis and medicinal chemistry. He then joined the group of Justin Lopchuk at H. Lee Moffitt Cancer Center and Research Institute, where he is now a Research Scientist, developing novel reagents and methods for the synthesis of pharmaceutically important functionality, and actively involved in the group's medicinal chemistry efforts. |

|
Yvonne "Eve" Hall completed her undergraduate studies at North Carolina State University with a Chemistry BS degree. She is currently pursuing her PhD under the guidance of Dr. Justin Lopchuk. Her research focuses on the development of methods and reagents for sulfur containing compounds important for drug discovery. |

|
Prakash K. Warghude was born in Maharashtra, India. He received his B.Sc. and M.Sc. degrees from ASC College, Rahuri. He obtained his Ph.D. in 2022 from IISER Pune under the supervision of Prof. Ramakrishna G. Bhat, where he worked on the synthesis of spiroxindoles. From 2022 to 2024, he conducted postdoctoral research with Prof. Fujie Tanaka at OIST, Japan, focusing on the synthesis of chiral diamine catalysts. In 2024, he joined the H. Lee Moffitt Cancer Center as a postdoctoral fellow in the laboratory of Prof. Justin Lopchuk. His current research interests include asymmetric sulfur chemistry and medicinal chemistry. |

|
Yun-Pu Chang received her B.S. and M.S. degrees in chemistry from National Taiwan Normal University, where she conducted research with Prof. Kwunmin Chen. She received her Ph.D. in chemistry at the University of California-Davis, working with Prof. Annaliese K. Franz, where she developed new chiral silanol ligands for enantioselective catalysis. She is currently a postdoctoral fellow at H. Lee Moffitt Cancer Center and Research Institute under the guidance of Prof. Justin M. Lopchuk, developing new methods for covalent inhibition. |

|
Taylor A. Stephens is from Tallahassee, FL and graduated with her B.S. degrees in chemistry and mathematics from the University of Florida, where she conducted research with Prof. Leslie Muray working on Fe coordination chemistry. She joined Tehshik Yoon's lab at the University of Wisconsin at Madison in 2024, where she is working on photochemical asymmetric dual catalytic reactions with Lewis acids. |

|
Tehshik Yoon is a Professor of Chemistry at the University of Wisconsin-Madison, where he has served on the faculty since 2005. He received his PhD from Caltech with David MacMillan (2002) and conducted postdoctoral research at Harvard with Eric Jacobsen. Tehshik has been on the faculty at UW-Madison since 2005. His research group has broad interests in the development of synthetically useful transformations promoted by visible light. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved