Org. Synth. 2026, 103, 283-300
DOI: 10.15227/orgsyn.103.0283
Photochemical Synthesis of Pyrroles from Furans and Amines
Submitting Authors: Jaehyun You, Donghyeon Kim, and Yoonsu Park*
Checking Authors: Eléonore Tacke, Bjarne T. Schulz and Nuno Maulide*
21. Procedure (Note 1)
A. 3-Phenylfuran (1). An 80 ℃ oven-dried 250-mL, three-necked, round-bottomed flask (24/40 joints) containing Teflon-coated magnetic stirring bar (4 cm, egg-shaped) were added 3-furanboronic acid (4.92 g, 44 mmol, 1.1 equiv.) (Note 2), Pd(PPh3)4 (1.85 g, 1.6 mmol, 0.04 equiv.) (Note 3) and Na2CO3 (9.33 g, 88 mmol, 2.2 equiv.) (Note 4). One side-neck of the flask was fitted with a PTFE-plug stopcock adapter connected to a Schlenk line. The central neck was fitted with an air-cooled reflux condenser (43.5 cm × 3.5 cm active area), which was capped with a rubber septum. The remaining neck was fitted with a rubber septum pierced by a thermometer. All ground-glass joints were sealed with vacuum grease, secured with Keck clips, and all septa were wrapped with Parafilm®. The assembled apparatus was purged by subjecting it to three evacuation/nitrogen backfill cycles (2 × 5 min, 1 × 10 min). A solvent mixture of toluene (60 mL) (Note 5), water (25 mL), and methanol (20 mL) (Note 6) was then added via 20-mL plastic syringes fitted with 21-gauge needles. Bromobenzene (6.28 g, 4.2 mL, 40 mmol, 1.0 equiv.) (Note 7) was added to the flask via a 10-mL plastic syringe fitted with a 23‐gauge needle (Figure 1B).

Figure 1. A. Reaction setup; B. Reaction mixture before stirring; C. Reaction mixture after 14 h of stirring (photos provided by the submitting authors)
The reaction mixture was heated to 80 ℃ (oil bath temperature) and stirred at 500 RPM for 14 h, maintaining a gentle reflux under a positive nitrogen pressure. The reaction progress was monitored by TLC analysis (Note 8). Upon completion, the heating bath was removed, and the flask was allowed to cool to room temperature (Figure 1C).
The cooled reaction mixture was vacuum filtered through a pad of Celite® (6 g) (Note 10, Figure 2A) on a 60 mL-sized sintered glass frit, and the filter cake was rinsed with dichloromethane (3 × 50 mL) (Note 11). The combined filtrate in a 500 mL-sized round-bottomed flask (Figure 2B) was transferred to a 1-L separatory funnel and washed with water (150 mL). The organic layer was separated (Figure 2C), and the aqueous phase was subsequently extracted with dichloromethane (3 × 100 mL). The combined organic layers were washed with brine (3 × 100 mL) (Note 12), dried over anhydrous Na2SO4 (10 g) (Note 13), and filtered through a 60 mL-sized sintered glass frit into a 1-L round-bottom flask. The organic layer was then concentrated by rotary evaporation in two stages: first at 35 ℃ (300 → 140 torr) to remove the dichloromethane, followed by heating at 50 ℃ (<90 torr) to remove toluene.

Figure 2. A. Celite filtration setup; B. Crude mixture after filtration; C. Crude mixture after transferring to a separatory funnel; D. Crude mixture after adding sodium sulfate (photos provided by the submitting authors)
The crude residue was dissolved in a minimal volume of dichloromethane (4 mL) and purified by flash column chromatography on silica gel (Notes 14, 15) (Figure 3A). This procedure afforded 3-phenylfuran 1 (3.92 g, 68% yield, 98.8% purity) as a white crystalline solid (Figure 3B) (Notes 16, 17).
Figure 3. A. Setup for flash column chromatography; B. Appearance of product (2) after purification (photos provided by the submitting authors)
B. 1-(1-Methyl-1-phenylethyl)-3-phenyl-1H-pyrrole (2). To a flame-dried, 250-mL, three-necked round-bottomed flask (24/40 joints) containing a Teflon-coated magnetic stir bar (4 cm, egg-shaped) were added 1 (1.73 g, 12.0 mmol, 1.00 equiv.) (Note 16), tris(pentafluorophenyl)borane (614.4 mg, 1.2 mmol, 0.10 equiv.) (Note 18), and Fukuzumi's catalyst (383.3 mg, 0.96 mmol, 0.1 equiv.) (Note 19). One side-neck of the flask was fitted with a PTFE-plug stopcock adapter connected to a Schlenk line. The central neck was fitted with an air-cooling reflux condenser (43.5 cm × 3.5 cm active area), which was capped with a rubber septum. The remaining neck was fitted with a rubber septum pierced by a thermometer. All ground-glass joints were sealed with vacuum grease, secured with Keck clips, and all septa were wrapped with Parafilm®. The assembled apparatus was purged by subjecting it to three evacuation/nitrogen backfill cycles (2 × 5 min, 1 × 10 min).
Under a positive nitrogen pressure, anhydrous dichloromethane (120 mL, Note 11) was added using six 20-mL plastic syringes fitted with 21‐gauge needles, followed by the addition of cumylamine (1.947g, 2.07 mL, 14.4 mmol, 1.2 equiv.) using a 5-mL plastic syringe fitted with 23-gauge needle (Figure 4A, Note 20). The resulting mixture was stirred (150 RPM) and irradiated for 24 h using two KessilTM PR-160L (456 nm) lamps positioned 6 cm from the flask and set to 100% intensity (Figure 4B). During irradiation, the internal temperature was observed to increase from 17 ℃ to 27 ℃.
Figure 4. A. Reaction mixture before irradiation; B. Reaction mixture during 456 nm Blue LEDs irradiation while stirring at room temperature; C. Reaction mixture after irradiation (photos provided by the submitting authors)
After 24 h, the irradiation was stopped (Note 21) (Figure 4C). The magnetic stir bar was removed, and the mixture was transferred to a 250-mL round-bottomed flask. The solvent was removed by rotary evaporation (35 ℃, 290 → 15 torr) to afford a brown, viscous oil. To ensure quantitative transfer, this oil was dissolved in dichloromethane (10 mL), transferred to a 16-mL glass vial, and concentrated again by rotary evaporation (35 ℃, 375 → 15 torr).
The crude material was dissolved in dichloromethane (8 mL) and subjected to flash column chromatography (Note 22) on silica gel for purification (Figure 5A). Elution with an isocratic eluent mixture of n-hexane/EtOAc (98:2) (Note 23) afforded 1-(1-methyl-1-phenylethyl)-3-phenyl-1H-pyrrole 2 (2.65 g, 85% yield, 98.3% purity) (Notes 24, 25) as a pale-yellow solid (Figure 5B).
Figure 5. A. Setup for flash column chromatography; B. Appearance of final product (2) after purification (photos provided by the submitting authors)
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regards to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/about/governance/committees/chemical-safety.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with (
Na2CO3,
Pd(PPh3)4,
toluene,
methanol,
n-hexane,
n-heptane,
dichloromethane,
ethyl acetate,
3-furanboronic acid,
3-phenylfuran,
bromobenzene, and
Fukuzumi's catalyst), as well as the proper procedures for the setup and operation of the Kessil 456 nm LED light source, including its installation and use, maintenance of adequate cooling to control reaction temperature during prolonged irradiation, shielding of the reaction apparatus to minimize exposure to direct or reflected light, and handling of reaction vessels under continuous irradiation.
2.
3-Furanboronic acid (99.63%) was purchased from BLDpharm and used as received.
3.
Pd(PPh3)4 (98%) was purchased from BLDpharm and used as received.
4. The submitting authors purchased
Na2CO3 (99.5%) from SAMCHUN and used as received. The checking authors purchased
Na2CO3 (99.5%) from Sigma-Aldrich and used as received.
5. The submitting authors purchased
toluene (With Sure Seal, anhydrous, 99.8%) from Sigma Aldrich and used as received. The checking authors purchased
toluene (AcroSeal®, 99.85%) from Thermo Scientific-Acros and used as received.
6. The submitting authors purchased
Methanol (With Sure Seal, Anhydrous, 99.8%) from Sigma-Aldrich and used as received. The checking authors purchased
Toluene (AcroSeal®, 99.85%) from Thermo Scientific Acros and used as received. Water in the reaction was deionized but not degassed prior to use.
7.
Bromobenzene (99.5%) was purchased from Sigma Aldrich and used as received.
8. The progress of the reaction was monitored by TLC analysis on silica gel with
n-hexane (
Note 9) used as an eluent. The checking authors used
n-heptane (
Note 9). R
f values using
n-heptane: The
bromobenzene starting material (
Br) has R
f 0.62, The other starting material
furan-3-boronic acid (
Fu) has R
f 0.06 and
3-phenylfuran in reaction crude mixture (
P) has R
f 0.31 (Figure 6). (
c indicates co-spots).
Figure 6. TLCs by checking authors (100% n-heptane) (photos provided by the checking authors)
9. The submitting authors purchased
n-hexane (95%, GR GRADE) from DUKSAN REAGENTS and used as received. The checking authors purchased
n-heptane (99.5%, AR grade) from Fischer Scientific and used as received.
10. The submitting authors purchased Celite (545 Filter, Practical GRADE) from DUKSAN REAGENTS and used as received. Checking authors purchased Celite (545 Filter) from Sigma-Aldrich.
11. The submitting authors purchased
dichloromethane (anhydrous, >99.8%, contains 40-150 ppm amylene as stabilizer) from Sigma Aldrich and used as received. The checking authors purchased
dichloromethane (ACS reagent, reag. ISO, ≥99.9% GC) and used as received.
12. Submitting authors purchased
sodium chloride (99.0%, Extra Pure grade) from SAMCUHN and used to make a saturated aqueous solution. Checking authors purchased
sodium chloride (99.5%) from Fischer Scientific and used as received.
13. The submitting authors purchased
sodium sulfate (anhydrous, 99.5%) from SAMCHUN and used as received. Checking authors purchased
sodium sulfate (99%) from Sigma Aldrich and used as received.
14. The submitting authors purchased Silica (SiO
2, Silica gel 60, Particle size: 0.040-0.063 mm) from Sigma Aldrich and used as received. Checking authors purchased Silica (SiO
2, Silica gel 60, Particle size: 0.040-0.063 mm) from Macherey-Nagel and used as received.
15. A 5-cm diameter glass column was packed with silica gel (130 g, silica height 16.5 cm) using a wet pack method with
n-heptane. The silica bed was topped with a protective layer of anhydrous
Na2SO4 (~1 cm). Fractions were collected in 250-mL Erlenmeyer flasks (8 fractions), and those containing the product (fractions 2-4, as determined by TLC, Figures 7; the checking authors observed a side-product eluting in fraction 4, which was not included in the pooled fractions) were combined in a 1-L round-bottomed flask. The bulk of the eluent was removed by rotary evaporation (35 ℃, 140 →100 torr). The resulting solid was quantitatively transferred to a 50-mL vial with
dichloromethane rinses (4 x 5 mL). After removing the rinse solvent under reduced pressure by rotary evaporation (35 ℃, 350 →140 torr), the product was further dried under vacuum (0.1 torr) at 22 ℃ for 14 h to yield the final compound.
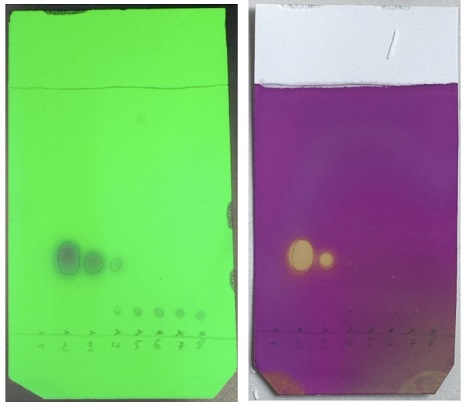
Figure 7. TLC (n-heptane/EtOAc (98:2)) of column fractions visualized using A) UV lamp (254 nm); B) KMnO4 staining agent (photos provided by the checking authors)
16. The product (
1) possesses the following properties: R
f = 0.31 (
n-heptane); m.p. = 57.4-58.2 ℃;
1H NMR
pdf (700 MHz, CDCl
3) δ 7.77 (s, 1H), 7.57 - 7.48 (m, 3H), 7.41 (m, 2H), 7.31 (m, 1H), 6.81 - 6.66 (m, 1H).
13C NMR
pdf (176 MHz, CDCl
3) δ 143.8, 138.6, 132.7, 128.9, 127.1, 126.6, 126.0, 109.0; IR(ATR) 3147, 3031, 1606, 1512, 1451, 1367, 1265, 1163, 1101, 752, 691; HRMS (GC-MS; EI) m/z: Calcd. for C
10H
8O [M]
+: 144.0570, found: 144.0566. The purity of
1 was determined to be 98.8% by qNMR using 1,3,5‐trimethoxybenzene (purity 98.0%, purchased from TCI, used as received) as an internal standard.
17. A second run performed on half scale (20 mmol) provided 1.96 g of product
1 (68% yield) in 98.9% purity as determined by quantitative
1H NMR
pdf (
Note 16). The submitting authors reported one full-scale run to yield 3.94 g (68% yield, 97.7% purity) and another identical run to yield 4.63 g (80% yield, 98.2% purity).
18.
Tris(pentafluorophenyl)borane (>98%) was purchased from TCI and used as received.
19.
9-Mesityl-10-methylacridinium tetrafluoroborate (>95%) was purchased from Sigma-Aldrich and used as received.
20.
Cumylamine (>98.0%) was purchased from TCI and used as received.
21. Upon 24 h irradiation, the conversion of the starting material was confirmed by TLC analysis on silica gel with
n-hexane/
EtOAc (98:2) used as an eluent. The checking authors used
n-heptane/
EtOAc (98:2). The plate was visualized using a UV (254 nm) irradiation, followed by staining with
KMnO4. Starting material-to-product ratios of 7:93, 4:96, and 3:97 were observed at 24, 36, and 48 h of reaction time, respectively, and the product
2 was observed at an R
f value of 0.20 (
n-hexane/
EtOAc (98:2), Figure 8) and 0.16 (
n-heptane/
EtOAc (98:2), Figure 11).
Figure 8. TLC (n-heptane/EtOAc (98:2)) of the reaction crude mixture after 24 h of irradiation (photos provided by the checking authors).
22. A 3.5-cm diameter glass column (35/20 joint) was wet-packed with silica gel (100 g) using 2%
EtOAc in
n-hexane as the eluent (
Note 23). The checking authors used a 3.5-cm diameter glass column that was wet-packed with silica gel (100 g, 31.5 cm silica height) using
n‐heptane/
EtOAc (98:2) as the eluent. The silica bed was topped with a protective layer of anhydrous
Na2SO4 (~1 cm). During elution, fractions were collected in 100 mL Erlenmeyer flasks, and those containing the product (fractions 6-8, as determined by TLC, Figure 9) were combined in a 1-L round-bottomed flask. The combined organic layer was concentrated by rotary evaporation (35 ℃, 140 →100 torr). The crude mixture was redissolved in
EtOAc (12 mL) and transferred to a 100-mL round-bottomed flask. The combined organic layer was concentrated by rotary evaporation (35 ℃, 110 →60 torr). After removing the rinse solvent under reduced pressure by rotary evaporation (35 ℃, 110 →60 torr), the product was further dried under vacuum (0.2 torr) at ambient temperature for 9 h to yield the final compound.

Figure 9. TLC (n-heptane/EtOAc (98:2)) of column fractions visualized using A) UV lamp (254 nm); B) KMnO4 staining agent (photos provided by the checking authors)
23. Submitting authors purchased
EtOAc (99.5%, Extra Pure GRADE) from DUKSAN REAGENTS and used as received. Checking authors purchased
EtOAc from Fischer Scientific (99%, LR grade) and used as received.
24. The product (
2) possesses the following properties: R
f = 0.16 (
n‐heptane/
EtOAc (98:2));
1H NMR
pdf (600 MHz, CDCl
3) δ 7.58 (dd,
J = 8.4, 1.3 Hz, 2H), 7.39 - 7.32 (m, 4H), 7.30 - 7.24 (m, 1H), 7.19 (tt,
J = 7.4, 1.2 Hz, 1H), 7.16 - 7.10 (m, 3H), 6.86 (t,
J = 2.6 Hz, 1H), 6.57 (dd,
J = 3.0, 1.9 Hz, 1H), 1.97 (s, 6H);
13C NMR
pdf (151 MHz, CDCl
3) δ 147.9, 136.1, 128.7, 128.6, 127.2, 125.3, 125.1, 125.0, 124.4, 120.4, 116.3, 106.0, 60.7, 30.6; mp = 95.2-96.1 ℃; HRMS (ESI-MS) m/z: Calcd. for C
19H
20N [M+H]
+: 262.1591, found: 262.1590. The purity of
2 was determined to be 98.3% by qNMR using
1,3,5-trimethoxybenzene (purity 98.0%, purchased from TCI, used as received) as an internal standard.
25. A second run performed on half scale (6 mmol) provided 1.26 g of product
2 (80% yield) in 99.4% purity as determined by quantitative
1H NMR
pdf (
Note 24). The submitting authors reported one full-scale run to yield 2.57 g, (82% yield, 99.2% purity) and another identical run to yield 2.39 g (76% yield, 99.3% purity).
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Skeletal editing technique allows a conversion of one heterocyclic scaffold to another, enabling an efficient access to a diverse library of compounds with precise control of atomic composition.
2, 3, 4 Given the impact of single atomic effects on compounds of medicinal interest,
5, 6 the ability to formally exchange a heteroatom within an aromatic ring is particularly appealing. Such methods would facilitate evaluation of structure-activity relationship and propel the expansion of chemical library that cannot be accessed by traditional approaches.
7, 8Despite these advantages, conventional skeletal editing methods often require harsh conditions and highly reactive reagents, dissuading chemists from the practice.
9, 10 In this regard, we recently reported a single-atom editing reaction of furan that operates under photocatalytic conditions, which enable a direct and selective exchange of the furan oxygen for a substituted nitrogen sourced from alkylamines, at ambient temperature.
11Figure 10. Substrate scope (amine)
To evaluate the synthetic applicability, an array of alkylamines was first subjected to the optimized catalytic conditions with 3-phenylfuran
1 as a model substrate (Figure 10). The desired reaction proceeded well regardless of the substitution patterns at the para-position of the benzylamine (
3-
7). Pharmaceutically relevant moieties, such as piperidine derivatives (
8) or tryptamine derivatives (
9), were amenable to the optimized conditions, highlighting the synthetic utility of the protocol.
12 Stereochemical integrity was confirmed by using enantiomerically enriched and diastereomeric amines, where chiral information was fully preserved throughout the product-forming processes (
10,
11). A free hydroxyl group was tolerated (
11,
12), and strained moieties (
13) or sterically hindered groups (
14 -
17) did not interfere with the desired reactivity, highlighting the synthetic usefulness of the protocols.
Figure 11. Substrate scope (furan)
Variations on the furanyl moiety were further examined (Figure 11). Unsubstituted furan, an abundant petroleum feedstock, was successfully converted to its pyrrole analog (18). Substitutions at the para position of the 3-phenyl moiety afforded good to excellent conversion to the pyrrole congeners (19-24), and both the electronic (21-24) and steric perturbations (20) were tolerated. The developed protocol can also be applied to furans containing a heteroaryl substituent (25) and a π-extended aromatic group (26). Furthermore, disubstituted (27-30) and trisubstituted (31) furans were compatible with the developed protocol.
Figure 12. Overall proposed mechanism of the photocatalytic furan-to-pyrrole conversion
The proposed mechanism pathway is shown in Figure 12. The reaction is initiated by reductive quenching of the excited-state photocatalyst (PC*) by furan (I) to generate furanyl radical cation II. The generated electrophilic radical II undergoes a facile C-N bond formation with a nucleophilic amine, followed by a spontaneous deprotonation under basic conditions. The resultant intermediate III proceeds with facile C-O bond cleavage for delocalizing electron density over the π-surface. Such driving force affords an oxidizing intermediate IV upon ring opening, followed by an oxidative quenching to furnish a neutral intermediate V. The neutral enamine-aldehyde-type intermediate V may undergo a Paal-Knorr-type condensation, giving the observed pyrrole product VI by releasing H2O as a byproduct.
In conclusion, we have developed a one-pot protocol that enables the direct conversion of furan to pyrrole under mild photocatalytic conditions on a 2-g scale. We expect that the succinct nature of the protocol would accelerate drug discovery campaigns by allowing direct access to a broad range of drug candidates in an efficient way.
Appendix
Chemical Abstracts Nomenclature (Registry Number)
3-Furanboronic acid: (furan-3-yl)boronic acid; (55552-70-0)
Bromobenzene; (108-86-1)
Na2CO3: Sodium carbonate; (497-19-8)
Pd(PPh3)4: Tetrakis(triphenylphosphine)palladium(0); (14221-01-3)
3-Phenylfuran (1); (13679-41-9)
Cumylamine: α,α-Dimethylbenzylamine; (585-32-0)
B(C6F5)3: Tris(pentafluorophenyl)borane; (1109-15-5)
Fukuzumi's catalyst: 9-Mesityl-10-methylacridinium tetrafluoroborate; (1442433-71-7)
1-(1-Methyl-1-phenylethyl)-3-phenyl-1H-pyrrole (2); (161768-23-6)
EtOAc: Ethyl acetate; (141-78-6)

|
Jaehyun You was born in Seoul, Korea, in 2000. He received his B.S. degree in chemistry from Korea Advanced Institute of Science and Technology (KAIST). He is now enrolled in an integrated M.S.-Ph.D. program at the Korea Advanced Institute of Science and Technology (KAIST), under the guidance of Professor Yoonsu Park, where his research focuses on the development of new photocatalytic skeletal editing reactions. |

|
Donghyeon Kim was born in Chuncheon, Korea, in 1996. He received his B.S. degree in chemistry from Sogang University (2022). He is now enrolled in an integrated M.S.-Ph.D. program at the Korea Advanced Institute of Science and Technology (KAIST), under the guidance of Professor Yoonsu Park. Currently, he is focused on the interconversion between heteroarene scaffolds, utilizing skeletal editing |

|
Yoonsu Park received his Ph.D. degrees in chemistry from Korea Advanced Institute of Science and Technology (KAIST) under the supervision of Prof. Sukbok Chang in 2019. After postdoctoral research with Prof. Paul J. Chirik at Princeton University, he began his independent career at his alma mater as an assistant professor in 2022. The Park group is interested in developing sustainable catalysis directed towards the synthesis of value-added compounds. |

|
Eléonore Tacke received her PhD degree in Organic Chemistry from the University Paris-Saclay (France) at the Institut de Chimie des Substances Naturelles (ICSN-CNRS) under the supervision of Dr. Arnaud Chevalier in 2024, where she developed multiple synthetic routes towards new hybrid fused fluorescent dyes. Currently, she is a postdoctoral researcher in Prof. Nuno Maulide's group at the University of Vienna. Her research interests span a wide range of synthetic methodologies, including heterocyclic chemistry and medicinal chemistry. |

|
Bjarne Schulz received his M.Sc. degree in Chemistry from the Technical University of Berlin in 2024, where he conducted research under the supervision of Prof. Martin Oestreich on the synthesis of chiral superacids and their application in superelectrophile catalysis. He is currently pursuing a Ph.D. in the group of Prof. Nuno Maulide in collaboration with Boehringer Ingelheim. His research focuses on synthetic methodologies and the development of novel, environmentally friendly drug derivatives. |

|
Nuno Maulide obtained his Ph.D. at the Université catholique de Louvain under the supervision of Prof. István E. Markó in 2007, followed by a postdoctoral stay in the group of Prof. Barry M. Trost at Stanford University. In 2009, he started his independent career at the Max-Planck Institut für Kohlenforschung. Following this, in 2013, he became a Full Professor at the University of Vienna. His research interests are broadly spread in the field of organic chemistry and include method development, total synthesis and medicinal chemistry-driven investigations. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved