Org. Synth. 2003, 80, 57
DOI: 10.15227/orgsyn.080.0057
(4S)-4-(1-METHYLETHYL)-5,5-DIPHENYL-2-OXAZOLIDINONE
[(2-Oxazolidinone, 4-(1-methylethyl)-5,5-diphenyl-, (4S)-)]
Submitted by Meinrad Brenner
1, Luigi La Vecchia
2, Thomas Leutert
2, and Dieter Seebach
1.
Checked by Mitsuru Kitamura and Koichi Narasaka.
1. Procedure
A. N-(tert-Butoxycarbonyl)-L-valine methyl ester(1) (Note 1). A 1-L, three-necked flask, fitted with a pressure-equalizing, 50-mL addition funnel, thermometer, and magnetic stirring bar is charged under an argon atmosphere (Note 2) with (S)-N-(tert-butoxycarbonyl)valine (65.2 g, 0.300 mol) (Note 3), dimethylformamide (DMF) (440 mL) (Note 4) and potassium hydrogen carbonate (60.1 g, 0.600 mol) (Note 5). Methyl iodide (29.9 mL, 0.480 mol) (Note 6) is added dropwise to this suspension over a period of 30 min. After the addition is complete, stirring is continued for 3 hr. The reaction mixture is poured into a 2-L separatory funnel, diluted with water (1.2 L) and extracted with a 1:1 mixture of ethyl acetate and hexane (3 × 250 mL). The organic phases are combined and successively washed with water (2 × 250 mL), aqueous 5% sodium sulfite (2 × 250 mL), and brine (250 mL). After drying over anhydrous magnesium sulfate, the solvent is removed and the oily residue is dried under vacuum (10 mbar) at room temperature. Crude 1 (69.4 g) is used in Step C without further purification.
B. Phenylmagnesium bromide. Under an argon atmosphere (Note 2) a 2-L, three-necked flask, fitted with a reflux condenser, pressure-equalizing, 500-mL addition funnel, thermometer, and magnetic stirring bar, is charged with magnesium turnings (25.5 g, 1.05 mol) and tetrahydrofuran (THF) (130 mL) (Note 7). Bromobenzene (15.7 g, 0.100 mol) (Note 8) is then added, and the reaction is initiated by warming with a heat gun. A solution of bromobenzene (149 g, 0.95 mol) in THF (500 mL) is added at such a rate that gentle refluxing is maintained. After the addition is complete (Note 9), the reaction mixture is heated to reflux for 1 hr with an oil bath.
C. (S)-[1-(Hydroxydiphenylmethyl)-2-methylpropyl]carbamic acid, tert-butyl ester(2). The Grignard solution obtained in Step B is cooled with an ice bath (Note 10) and a solution of N-(tert-butoxycarbonyl)-L-valine methyl ester (1) (69.4 g, 0.30 mol) in THF (300 mL) is added at a rate to maintain the internal temperature below 6°C (Note 11). After the addition is complete, the reaction mixture is allowed to warm to room temperature and stirring is continued for another 15 hr (Note 12).
The reaction mixture is poured into an ice-cold aqueous saturated ammonium chloride solution (700 mL). The aqueous phase is separated and extracted with ethyl acetate (2 × 500 mL). The organic phases are combined, washed with brine (500 mL), and dried over anhydrous magnesium sulfate. After removal of the solvent under reduced pressure, crude 2 (115 g) is obtained as a white solid.
The crude product is dissolved in boiling ethyl acetate (600 mL) (Note 13) in a 1-L flask fitted with a reflux condenser and a magnetic stirring bar; then hexane (230 mL) is added. A few minutes after cessation of heating, a white solid starts to precipitate. The suspension is allowed to cool to room temperature, then is cooled for an additional hour with an ice bath. The solids are collected, washed with ice-cold hexane (100 mL), and dried under high vacuum (0.1 mbar) at room temperature for 12 hr to give purified 2 (77.1 g, 72%) (Note 14) as a white solid. The mother liquor is concentrated under reduced pressure (to 220 g) and hexane (150 mL) is added. A few minutes after the addition of hexane, colorless crystals start to form. After standing for several hours at room temperature, the crystals are collected, washed with ice-cold hexane (100 mL) and dried as described above to yield a second crop of 2 (12.5 g, 12%). A third crop (11.3 g, 11%) is obtained after concentration (to 70 g) and treating of the mother liquor as described above (Note 15).
D. (4S)-4-(1-Methylethyl)-5,5-diphenyl-2-oxazolidinone (3). Under an argon atmosphere (Note 2), a 2-L, three-necked flask, fitted with a thermometer and magnetic stirring bar (Note 16) is charged with (S)-[1-(hydroxydiphenylmethyl)-2-methylpropyl]carbamic acid, tert-butyl ester (2) (97.8 g, 0.275 mol) (Note 17) and THF (1.8 L). The resulting solution is cooled to an internal temperature below 5°C with an ice bath. Potassium tert-butoxide (37.0 g, 0.330 mol) (Note 18) is then added in one portion (Note 19). A few minutes after the addition of potassium tert-butoxide a white solid begins to precipitate. Stirring below 5°C is continued for 2 hr. The resulting suspension is poured into a 10% aqueous solution of ammonium chloride (2 L) and stirred for 10 min. The white solids are collected and washed with water (4 × 400 mL) (Note 20). The solids are transferred to a 1-L flask fitted with a reflux condenser and a magnetic stirring bar. Methanol (650 mL) is added and the resulting suspension is heated to reflux for 1 hr with stirring. The mixture is allowed to cool to room temperature and stirring is continued for 1 hr in an ice bath. The white solids are collected and washed with ice-cold methanol (100 mL). After drying for 24 hr under high vacuum (0.1 mbar) at room temperature, oxazolidinone 3 (72.5 g, 94%) (Note 21) is obtained as a white solid.
2. Notes
1.
N-(tert-Butoxycarbonyl)-L-valine methyl ester is also commercially available (Aldrich Chemical Co., Inc). The procedure described here is taken from the literature
3 and is readily reproducible.
2.
The flask was flushed with
argon and the
argon atmosphere was maintained with an
argon balloon.
3.
(S)-N-(tert-Butoxycarbonyl)valine (puriss grade) was obtained from Fluka Chemie AG. (the submitters) or from Tokyo Chemical Industry (the checkers).
4.
DMF (p. a. grade) was used as received from Merck AG. The checkers obtained
DMF (99%) from Kokusan Chemical Co., Ltd.5.
Powdered
potassium hydrogen carbonate (purum grade) was purchased from Fluka Chemie AG (the submitters) or Kokusan Chemical Co., Ltd. (the checkers).
6.
Methyl iodide (purum grade) was obtained from Fluka Chemie AG (the submitters) or Tokyo Chemical Industry (the checkers).
7.
THF (p. a. grade) was used as received from Merck AG. The checkers obtained
THF from Kanto Chemical Co. (<0.005% water).
8.
Bromobenzene (puriss grade) was obtained from Fluka Chemie AG (the submitters) or Tokyo Chemical Industry (the checkers).
9.
The
bromobenzene solution is added over ca. 1.5 hr.
10.
The clear dark Grignard solution becomes a suspension after cooling with an ice bath.
11.
The protected valine ester is added over ca. 1.5 hr.
12.
TLC analysis indicates almost complete reaction after stirring for 3 hr at room temperature. Stirring is continued overnight to ensure complete reaction.
13.
The resulting light yellow solution is slightly turbid.
14.
The product exhibits the following properties:
mp 188.5-189°C.
[α]Dr.t. −65 (c 1.0, CH2Cl2) [The checkers obtained
[α]D29°C −60 (c 1.0, CH2Cl2)]. IR (CHCl
3) cm
−1: 3446, 2965, 1703, 1498, 1368.
1H NMR (400 MHz, CDCl
3) δ: 0.88 (d, J = 6.8, Me), 0.90 (d, J = 6.9, Me), 1.32 (s, t-Bu), 1.43 (br. s, t-Bu), 1.80 (septd, J = 6.8, 2.2, CH), 2.63 (s, OH), 4.42-4.50 (m, CH), 4.58-4.62 (m, CH), 5.01 (d, J = 10.2, NH), 7.13-7.32 (m, 6 H), 7.44-7.55 (m, 4 H).
13C NMR (100 MHz, CDCl
3) δ: 17.4, 22.7, 28.3, 28.8, 59.1, 79.0, 82.4, 125.3, 125.7, 126.7, 126.8, 128.2, 128.3, 145.6, 146.3, 156.3. Exact mass calc. for C
22H
29NO
3Na
+ (M + Na
+): 378.2040. Found (MALDI) m/z 378.2042. For comparison with literature data, see Ref.
4 and
5.
15.
The third crop contains
(4S)-4-(1-methylethyl)-5,5-diphenyloxazolidin-2-one (
3) as a byproduct (ca.
7 mol% as determined from the
1H NMR spectrum).
16.
Since the reaction mixture becomes a viscous suspension, a
large magnetic stirring bar or
mechanical stirrer has to be used.
17.
The three crops obtained in Step B are used.
18.
Potassium tert-butoxide (purum grade) was purchased from Fluka Chemie AG.
19.
The temperature rises to 8°C after the addition of
potassium tert-butoxide.
20.
The third and fourth washes are negative when tested for chloride with
silver nitrate.
21.
The enantiomer ratio was determined by HPLC (column: Chiralcel OD-H, 4.6 × 15 mm, 5 μm, Daicel Chemical Industries; eluent: hexane/2-PrOH 10:1; flow: 1 mL/min; detection: UV at 254 nm; (S)-
3: t
R = 4.3 min; (R)-
3: t
R = 12.8 min) to be >99:1. The product exhibits the following properties:
mp 253.5-254°C,
[α]Dr.t. −244 (c 0.61, CH2Cl2). IR (CHCl
3) cm
−1: 3460, 2967, 1759.
1H NMR (400 MHz, CDCl
3) δ: 0.69 (d, J = 6.6, Me), 0.90 (d, J = 7.0, Me), 1.81-1.90 (m, CH), 4.36 (d, J = 3.7, CH), 6.61 (br. s, NH), 7.23-7.40 (m, 8 H), 7.52-7.55 (m, 2 H).
13C NMR (100 MHz, CDCl
3) δ: 15.6, 20.8, 29.6, 65.9, 89.4, 125.7, 126.3, 127.7, 128.1, 128.2, 128.5, 139.2, 143.9, 158.8. Exact mass calc. for C
18H
19NO
2Na
+ (M + Na
+): 304.1308. Found (MALDI) m/z 304.1308. For comparison with literature data see Ref.
5,
6,
7,
8.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
The present procedure for the preparation of oxazolidinone
3 is a variation of the procedures described by Luche
4 and Davies.
5 Yields have been substantially enhanced by improving the purification procedures. The preparation of
3 starting from valine methyl or ethyl ester hydrochloride has been described by several authors.
6,7,8,9 These procedures suffer from moderate yields for the Grignard addition step and some of them use hazardous reagents like
phosgene.
Oxazolidinones, such as
4 and
5, have been introduced by Evans as very useful chiral auxiliaries.
10 These compounds are prepared from readily available chiral amino alcohols and are successfully employed in a variety of different stereoselective reactions.
11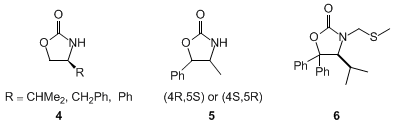
(4S)-4-(1-Methylethyl)-5,5-diphenyl-2-oxazolidinone (
3), whose preparation is described here, has several advantages over Evans' original auxiliaries:
8 i) Derivatives of
3 are more likely to crystallize. In many cases the separation and purification of diastereoisomers can be achieved by simple recrystallization rather than by expensive and time-consuming chromatography. ii) Acylation of
3 can be carried out at 0°C (instead of −78°C for
4 and
5) by deprotonation with BuLi, followed by treatment with an activated carboxylic acid derivative. iii) Lithium enolates of N-acyl derivatives of
3 can be obtained directly by treatment with BuLi at −78°C, in comparison to
4 and
5 when the more expensive
lithium diisopropylamide or
lithium hexamethyldisilazane is required. iv) The N-acyl derivatives of
3 can be cleaved by using
sodium hydroxide without any detectable nucleophilic attack on the oxazolidinone carbonyl group. Expensive and hazardous (in large scale) LiOH/H
2O
2 has to be used for the cleavage of N-acyl derivatives of
4 and
5. v) Due to its low solubility in most solvents, oxazolidinone
3 is easily recovered in high purity by simple filtration after the cleavage step.
Enolates of N-acyl derivatives of oxazolidinone
3 have been used for asymmetric alkylations,
5,7,8,9 aldol reactions,
8 Mannich reactions,
8 Michael additions to nitroolefins,
8,12 azide transfer reactions,
9 and samarium-Reformatsky reactions.
13 Furthermore, α,β-unsaturated N-acyl derivatives of
3 have been employed in Diels-Alder reactions and in Michael addition reactions of cuprates.
8 The N-methyl-thiomethyl derivative
6 of oxazolidinone
3 can be lithiated to provide a reagent which is synthetically equivalent to a chiral formyl anion.
14
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
(S)-4-(1-Methylethyl)-5,5-diphenyloxazolidin-2-one:
2-Oxazolidinone, 4-(1-methylethyl)-5,5-diphenyl-, (4S)- (9); (184346-45-0)
N-(tert-Butoxycarbonyl)-L-valine methyl ester:
Valine, N-[(1,1-dimethylethoxy)carbonyl]-, methyl ester, (S)- (9); (58561-04-9)
(S)-N-(tert-Butoxycarbonyl)valine:
L-Valine, N-[(1,1-dimethylethoxy)carbonyl]- (9); (13734-41-3)
Methyl iodide:
Methane, iodo- (8, 9); (74-86-4)
Phenylmagnesium bromide:
Magnesium, bromophenyl- (8, 9); (100-58-3)
Bromobenzene:
Benzene, bromo- (8, 9); (100-86-1)
Magnesium (8, 9); (7439-95-4)
Potassium tert-butoxide:
2-Propanol, 2-methyl-, potassium salt (9); (865-47-4)
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved