Org. Synth. 2003, 80, 160
DOI: 10.15227/orgsyn.080.0160
GENERATION AND [2+2] CYCLOADDITIONS OF THIO-SUBSTITUTED KETENES: trans-1-(4-METHOXYPHENYL)-4-PHENYL-3-(PHENYLTHIO)AZETIDIN-2-ONE
[(2-Azetidinone, 1-(4-methoxyphenyl)-4-phenyl-3-(phenylthio)-, trans-)]
Submitted by Rick L. Danheiser, Iwao Okamoto, Michael D. Lawlor, and Thomas W. Lee.
1.
Checked by Scott E. Denmark and Michael H. Ober.
1. Procedure
Caution! Diazo compounds are presumed to be toxic and potentially explosive and therefore should be handled with caution in a fume hood. Although in carrying out this reaction numerous times we have never observed an explosion, we recommend that these reactions be conducted behind a safety shield.
A. N-Benzylidene-p-anisidine. A 100-mL, three-necked, round-bottomed flask is equipped with an argon inlet adapter, rubber septum, glass stopper, and a magnetic stirring bar (Note 1). The flask is charged with 45 mL of dichloromethane (CH2Cl2) (Note 2) and 3.00 mL (0.030 mol) of benzaldehyde (Note 3), and then is cooled in an ice-water bath while a solution of 3.50 g (0.028 mol) of p-anisidine (Note 4) in 5 mL of CH2Cl2 is added dropwise via syringe over 15 min. After 30 min, 7.5 g of anhydrous magnesium sulfate is added in one portion. The ice-water bath is removed, and the reaction mixture is stirred at room temperature for 2 hr. The resulting mixture is then filtered through a sintered glass funnel with the aid of two 5-mL portions of CH2Cl2, and the filtrate is concentrated at reduced pressure by rotary evaporation at room temperature to afford a pale brown powder. This material is dissolved in 150 mL of ethanol heated in an 80°C water bath while 270 mL of hot water is added with stirring. The resulting solution is allowed to cool to room temperature and then is cooled in an ice-water bath for 2 hr. Filtration provides 5.31 g (88%) of N-benzylidene-p-anisidine as brown metallic plates (Note 5).
B.
S-Phenyl diazothioacetate. A
250-mL, three-necked, round-bottomed flask is equipped with a
magnetic stirring bar,
argon inlet adapter,
rubber septum, and a
50-mL pressure-equalizing dropping funnel fitted with a rubber septum (Note 1). The flask is charged with
50 mL of dry tetrahydrofuran (THF) (Note 6) and
12.0 mL (0.057 mol) of 1,1,1,3,3,3-hexamethyldisilazane (Note 7), and then is cooled in an ice-water bath while
20.7 mL (0.052 mol) of a 2.58M solution of n-butyllithium in hexane (Note 8) is added dropwise over 5 min by syringe. After 10 min, the resulting solution is cooled at −78°C in a
dry ice-acetone bath, and a solution of
7.00 mL (0.052 mol) of S-phenyl thioacetate (Note 9) in
40 mL of dry tetrahydrofuran is added dropwise via the addition funnel over 30 min (the funnel is rinsed with two 2-mL portions of dry THF). The reaction mixture is allowed to stir for 30 min at −78°C, and then
8.50 mL (0.063 mol) of 2,2,2-trifluoroethyl trifluoroacetate (Note 10) is added rapidly in one portion via syringe. After 10 min, the reaction mixture is poured into
200 mL of 5% aqueous hydrochloric acid and is extracted with three portions
(250, 100, and 100 mL) of ether. The combined organic phases are washed with
100 mL of saturated sodium chloride solution, dried over
anhydrous sodium sulfate, filtered, and concentrated under reduced pressure with a rotary evaporator, then under high vacuum to afford a colorless solid. This material is immediately dissolved in
100 mL of acetonitrile (Note 11) and is transferred into a
500-mL, three-necked, round-bottomed flask equipped with a
magnetic stirring bar,
argon inlet adapter,
rubber septum, and
125-mL pressure-equalizing dropping funnel fitted with a rubber septum. Water (0.95 mL, 0.053 mol) and
10.0 mL (0.072 mol) of triethylamine (Note 12) are added via syringe, and a solution of
26.9 g (0.076 mol) of dodecylbenzenesulfonyl azide2 (Note 13) in
45 mL of acetonitrile is then added through the dropping funnel over 15 min. After stirring at room temperature for 16 hr, the solution is concentrated at reduced pressure with a rotary evaporator and then a
vacuum pump to give a brown oil. Silica gel (20 g) is added, and the resulting slurry is loaded onto a column (60 mm) of 200 g of silica gel (230-400 mesh) and is eluted with
5% ethyl acetate-hexane to afford
7.16 g (
77%) of
S-phenyl diazothioacetate as a pale yellow oil
(Note 14).
C. trans-1-(4-Methoxyphenyl)-4-phenyl-3-(phenylthio)azetidin-2-one. A 500-mL, three-necked, round-bottomed flask is equipped with a magnetic stirring bar, reflux condenser fitted with an argon inlet adapter, glass stopper, and 50-mL, pressure-equalizing dropping funnel fitted with a glass stopper (Note 1). The flask is charged with 3.50 g (0.016 mol) of N-benzylidene-p-anisidine, 200 mL of CH2Cl2 (Note 2), and 0.045 g (0.10 mmol) of rhodium(II) acetate dimer (Note 15), and the resulting green solution is heated at reflux while a solution of 4.43 g (0.025 mol) of S-phenyl diazothioacetate in 40 mL of CH2Cl2 is added via the dropping funnel over 1 hr (the dropping funnel is rinsed with 2 mL of CH2Cl2). The reaction mixture is further heated at reflux for 5 min, then allowed to cool to room temperature. After transfer to a 500-mL, round-bottomed flask, the mixture is concentrated by rotary evaporation at reduced pressure to provide a brown oil. This material is filtered through a column of 50 g of silica gel (elution with 750 mL of CH2Cl2) to remove polar impurities and is concentrated under reduced pressure to afford a brown solid, which is washed on a sintered glass funnel with 10 mL of ethyl acetate and then 20 mL of hexane. The resulting pale yellow powder is dissolved in 40 mL of ethyl acetate at 80°C, and 400 mL of hexane (pre-heated in a water bath at 80°C) is then added in one portion. The resulting solution is allowed to cool to room temperature and then is cooled further at −20°C for 2 hr to afford 5.50 g (91%) of trans-1-(4-methoxyphenyl)-4-phenyl-3-(phenylthio)azetidin-2-one as off-white crystals (Notes 16 and 17).
2. Notes
1.
The apparatus is flame-dried under reduced pressure and then maintained under an atmosphere of
argon during the course of the reaction.
2.
Dichloromethane was distilled from calcium hydride immediately before use.
3.
Benzaldehyde was purchased from Aldrich Chemical Company, Inc., and was distilled before use.
4.
p-Anisidine was purchased from Aldrich Chemical Company, Inc. and used as received.
5.
The imine has the following physical properties:
mp 70-71°C (lit.
3 70-71°C);
1H NMR (500 MHz, CDCl
3) δ: 3.83 (s, 3 H), 6.93 (d, 2 H, J = 9.0), 7.24 (d, 2 H, J = 8.8), 7.46 (m, 3 H), 8.48 (s, 1 H), 7.89 (m, 2 H);
13C NMR (166 MHz, CDCl
3) δ: 55.7, 114.6, 122.4, 128.8, 128.9, 131.3, 136.6, 145.2, 158.5, 158.7; IR (CCl
4) cm
−1: 3065, 3030, 3002, 2959, 2935, 2909, 2880, 2835, 1626, 1529, 1505, 1464, 1366, 1289, 1245, 1191, 1106, 1040, 920, 882, 828, 761, 691, 542; Anal. Calcd for C
14H
13NO: C, 79.59; H, 6.20; N, 6.63. Found: C, 79.50; H, 6.05; N, 6.74.
6.
Tetrahydrofuran was distilled from sodium benzophenone ketyl immediately before use.
7.
1,1,1,3,3,3-Hexamethyldisilazane was purchased from Aldrich Chemical Company, Inc., and was distilled from
calcium hydride prior to use.
8.
n-Butyllithium was purchased from Fisher Scientific Company, and was titrated prior to use according to the method of Watson and Gilman.
49.
S-Phenyl thioacetate was purchased from Aldrich Chemical Company, Inc. and used as received.
10.
2,2,2-Trifluoroethyl trifluoroacetate was purchased from Aldrich Chemical Company, Inc., and was distilled from
calcium hydride prior to use.
11.
Acetonitrile was distilled from calcium hydride prior to use.
12.
Triethylamine was purchased from EM Science, and was distilled from calcium hydride prior to use.
13.
Dodecylbenzenesulfonyl azide was purchased by the checkers from TCI Americas Inc. and was used as received.
14.
S-Phenyl diazothioacetate decomposes slowly on storage at −20°C with the generation of
nitrogen gas. The diazo compound is best used immediately in the next step or purified by chromatography immediately before use. Physical properties are as follows:
1H NMR (500 MHz, CDCl
3) δ: 5.25 (s, 1 H), 7.40-7.45 (m, 3 H), 7.49-7.51 (m, 2 H);
13C NMR (166 MHz, CDCl
3) δ: 54.5, 127.6, 129.6, 130.1, 135.6, 184.3; IR (thin film) cm
−1: 3099, 3077, 2466, 2354, 2269, 2108, 1641, 1477, 1441, 1334, 1139, 1022, 854, 748, 690, 638.
15.
Rhodium(II) acetate dimer was purchased from Alfa Aesar Company and used as received.
16.
Physical properties of the β-lactam are as follows:
mp 144-146°C (lit.
142°C5,
147-148°C6);
1H NMR (500 MHz, CDCl
3) δ: 3.72 (s, 3 H), 4.25 (d, 1 H, J = 2.4), 4.77 (d, 1 H, J = 2.4), 7.52-7.51 (m, 2 H), 7.36-7.34 (m, 3 H), 7.31-7.26 (m, 5 H), 7.16 (d, 2 H, J = 9.2), 6.75 (d, 2 H, J = 9.2),;
13C NMR (166 MHz, CDCl
3) δ: 162.9, 156.5, 136.6, 132.5, 130.9, 129.6, 129.5, 129.4, 129.2, 128.2, 126.3, 118.8, 114.5, 63.3, 61.7, 55.6; IR (CCl
4) cm
−1: 3065, 3033, 3003, 2953, 2934, 2909, 2835, 1760, 1512, 1455, 1381, 1248, 1179, 1137, 1067, 1040, 828, 736, 696, 590; Anal. Calcd for C
22H
19NO
2S: C, 73.10; H, 5.30; N, 3.88; S, 8.87. Found: C, 73.03; H, 5.29; N, 4.02; S, 9.20.
17.
Cleavage of the p-methoxyphenyl group can be achieved in
65% yield by the following procedure. A
three-necked, 250-mL, round-bottom flask, equipped with a
magnetic stirbar,
rubber septum,
argon inlet adapter, and
glass stopper, is charged with
0.512 g (1.42 mmol) of trans-1-(4-methoxyphenyl)-3-(phenylthio)azetidin-2-one and
55 mL of acetonitrile. After cooling the flask in an
ice-water bath, a solution of
2.380 g (4.34 mmol) of ceric ammonium nitrate in 20 mL of water is added by cannula over 5 min followed by stirring for 1 hr. The reaction mixture is poured into 500 mL of water and is extracted with three
200-mL portions of ethyl acetate. The organic phases are combined and washed with
400 mL of 5% aqueous NaHCO3 solution. The washes are back extracted with
100 mL of ethyl acetate. The combined organic phases are washed additionally with six
200-mL portions of saturated aqueous Na2SO3 solution, then two
150-mL portions of saturated aqueous NaCl solution. After drying with Na
2SO
4, the solution is concentrated to give a brown oil. Silica gel (1.5 g) is added, and the resulting slurry is applied to a column of 10 g of silica gel and eluted with
10% ethyl acetate-hexane. The eluent is concentrated to provide a yellow solid which is dissolved in
125 mL of boiling hexane, which is then reduced to a total volume of 50 mL. The resulting solution is allowed to cool to room temperature, then is cooled further to −20°C to afford
0.234 g (
65%) of
4-phenyl-3-(phenylthio)-2-azetidinone as a pale yellow solid,
mp 111-112°C (lit.
7 110.5-111.5°C).
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
[2+2] Cycloadditions of ketenes with alkenes and alkynes constitute the most popular method for the synthesis of cyclobutanones and cyclobutenones. Unfortunately, however, this process is truly general only for highly nucleophilic ketenophiles such as conjugated dienes and enol ethers. In general, unactivated alkenes and alkynes fail to react in good yield with either alkyl- or aryl-substituted ketenes, or with ketene itself. To circumvent this limitation, dichloroketene is usually employed as a ketene equivalent, since this electrophilic ketene reacts well with many types of unactivated multiple bonds, and the resultant cycloadducts undergo facile dechlorination under mild conditions.
8The procedure described here illustrates a practical and convenient method for the generation of thio-substituted ketenes which participate in surprisingly facile cycloadditions with both activated and unactivated alkenes, alkynes, and imines to form four-membered carbocycles and heterocycles. As outlined in the following scheme, exposure of α-diazo thiol esters to the action of catalytic rhodium(II) acetate leads to a remarkably facile "thia-Wolff rearrangement," producing thio-substituted ketenes which combine with a variety of ketenophiles to provide access to α-thiocyclobutanones, cyclobutenones, and β-lactams.
As illustrated below, desulfurization of α-thiocyclobutanone cycloadducts is easily achieved in high yield upon exposure to either tributyltin hydride or activated zinc dust, and the overall sequence thus represents a useful new alternative to the existing dichloroketene-based methodology for the synthesis of a variety of four-membered carbocycles and heterocycles
9.
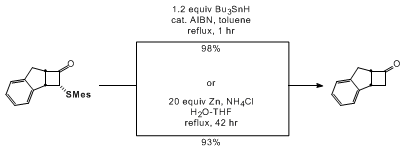
The requisite α-diazo thiol esters are conveniently prepared by using the "detrifluoroacetylative" diazo transfer strategy previously developed in our laboratory.
10 Cycloadditions are best carried out by using as little as 0.006 equiv of
rhodium(II) acetate to promote the thia-Wolff rearrangement. Reactions involving the more nucleophilic ketenophiles proceed smoothly in refluxing
dichloromethane (40°C), while cycloadditions with less reactive partners are best accomplished in
1,2-dichloroethane (83°C). As is standard for ketene cycloadditions, the optimal protocol involves slowly adding a solution of the diazo thiol ester to a solution of the ketenophile and catalyst in order to minimize competitive ketene dimerization.
Examples of the application of this chemistry to the preparation of cyclobutanones, cyclobutenones, and β-lactams are presented in the Table. The mesityl thiol ester has proven to be particularly effective in reactions with less ketenophilic alkenes, although with the more reactive ketenophiles nearly identical results are obtained using either the mesityl α-diazo thiol ester or the more readily available thiophenyl ester. In the case of readily available ketenophiles, the reaction is best conducted using excess alkene, alkyne, or imine, but in other cases the cycloaddition can be carried out with excess diazo thiol ester. The efficiency of the reaction with unactivated alkenes is especially notable, and compares favorably with results obtained previously employing dichloroketene. For example, addition of
dichloroketene to
methylenecyclohexane is reported to proceed in 55% yield,
11 while up to 81% of the desired [2+2] cycloadduct is produced in the reaction of (mesitylthio)ketene with this olefin under our conditions.
TABLE
Preparation of cyclobutanones and β-Lactams from α-Diazo Thiol Esters
|
|
|
aThe diazo thiol ester is treated with 0.02 equiv of Rh2(OAc)4 and 3.5-10 equiv of ketenophile. Procedure A: reaction in 1,2-dichloroethane at 83 °C for 3 h. Procedure B: reaction in dichloromethane at 40 °C for 3 h.
|
bIsolated yield and ratio of diastereoisomers.
|
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
trans-1-(4-Methoxyphenyl)-4-phenyl-3-(phenylthio)azetidin-2-one:
2-Azetidinone, 1-(4-methoxyphenyl)-4-phenyl-3-(phenylthio)-, trans- (9); (94612-48-3)
Benzaldehyde (8,9); (100-52-7)
p-Anisidine (8);
Benzenamine, 4-methoxy- (9); (104-94-9)
N-Benzylidene-p-anisidine:
Benzenamine, 4-methoxy-N-(phenylmethylene)- (9); (783-08-4)
1,1,1,3,3,3-Hexamethyldisilazane:
Silanamine, 1,1,1-trimethyl-N-(trimethylsilyl)- (9); (999-97-3)
Butyllithium:
Lithium, butyl- (8,9); (109-72-8)
S-Phenylthioacetate:
Ethanethioic acid, S-phenyl ester (9); (934-87-2)
2,2,2-Trifluoroethyl trifluoroacetate:
Acetic acid, trifluoro-, 2,2,2-trifluoroethyl ester (8,9); (407-38-5)
Triethylamine (8);
Ethanamine, N,N-diethyl- (9); (121-44-8)
4-Dodecylbenzenesulfonyl azide:
Benzenesulfonyl azide, 4-dodecyl- (13); (79791-38-1)
S-Phenyldiazothioacetate:
Ethanethioic acid, diazo-S-phenyl ester (13); (72228-26-3)
Rhodium(II) acetate dimer:
Acetic acid, rhodium(2+) salt (8,9); (5503-41-3)
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved