Org. Synth. 2005, 81, 77
DOI: 10.15227/orgsyn.081.0077
SYNTHESIS OF N-(tert-BUTOXYCARBONYL)-β-IODOALANINE METHYL ESTER: A USEFUL BUILDING BLOCK IN THE SYNTHESIS OF NONNATURAL α-AMINO ACIDS VIA PALLADIUM CATALYZED CROSS COUPLING REACTIONS
[L-Alanine, N-[(1,1-dimethylethoxy)carbonyl]-3-iodo-, methyl ester]
Submitted by Richard F. W. Jackson and Manuel Perez-Gonzalez
1.
Checked by Rick L. Danheiser and Aimee L. Crombie.
1. Procedure
A. N-(tert-Butoxycarbonyl)-O-(p-toluenesulfonyl)-L-serine methyl ester (2). A one-necked, 500-mL, round-bottomed flask equipped with a rubber septum and magnetic stirbar was charged with 26.1 g (119 mmol) of N-(tert-butoxycarbonyl)-L-serine methyl ester (1) (Note 1) and 200 mL of CH2Cl2 (Note 2). The solution is cooled in an ice bath at 0°C while 0.700 g (6.0 mmol) of 4-dimethylaminopyridine (4-DMAP), 1.1 g (12 mmol) of Me3NHCl (Note 3), and 22.7 g (119 mmol) of freshly recrystallized p-toluenesulfonyl chloride (TsCl) (Note 4) are added. The septum is replaced with a dropping funnel charged with 17 mL (119 mmol) of triethylamine (Et3N) in 50 ml of CH2Cl2 which is added dropwise to the reaction mixture at 0°C over 40 min (Note 5). The resulting slurry is stirred at 0°C for 2 hr and then poured into a mixture of 100 mL of ice, 100 mL of water, and 50 mL of 2M HCl solution. The aqueous layer is extracted with 100 mL of CH2Cl2, and the combined organic layers are washed with two 60-mL portions of brine, dried over magnesium sulfate, and concentrated by rotary evaporation to yield 59.4 g of a light yellow solid. This product may contain ca. 15% of starting material and TsCl that can be efficiently removed by crystallization according to the following procedure. The solid is dissolved in 140 mL of hot diethyl ether, filtered, and the filtrate is allowed to cool to room temperature and then to 0°C. Once crystallization begins (Note 6), a total of 250 mL of petroleum ether is added in five portions over 2 hr and then crystallization is allowed to proceed at −20°C overnight. The crystals are collected by suction filtration on a Büchner funnel and air-dried to give 28.3-30.8 g (64-69%) of 2 as a white solid (Note 7).
B. N-(tert-Butoxycarbonyl)-β-iodoalanine methyl ester (3). A one-necked, 250-mL, round-bottomed flask equipped with a rubber septum and magnetic stirbar is charged with 27.8 g (74.0 mmol) of N-(tert-butoxycarbonyl)-O-(p-toluenesulfonyl)-L-serine methyl ester (2) and 160 mL of acetone (Note 8). The solution is stirred at room temperature and 13.4 g (89.0 mmol) of NaI (Note 9) is added in one portion. The reaction mixture is stirred in the dark for 3 days, after which an additional 3.3 g (22 mmol) of NaI is added and stirring is continued for an additional day (Note 10). The reaction mixture is then suction filtered through a sintered glass funnel and the filtrate is collected in a one-necked, 500-mL, round-bottomed flask. The solid is washed with acetone until it is colorless. The solid is discarded and the filtrate is concentrated by rotary evaporation under reduced pressure (Note 11). The residual yellow oil is partitioned between 150 mL of diethyl ether and 60 mL of 1M sodium thiosulfate (Na2S2O3) solution. The organic layer is separated and washed with 40 mL of 1M Na2S2O3 50 mL of brine, dried over magnesium sulfate, filtered, and concentrated by rotary evaporation (Note 11) to afford 23.2 g of a colorless oil that solidifies on standing at 0°C. The solid is dissolved in 30 mL of hot (40°C) petroleum ether (bp 35-45°C), cooled to room temperature and then to 0°C. Once a precipitate appears (Note 6), the mixture is cooled at −20°C for 1 hr and the white solid is collected on a Büchner funnel and washed with cold petroleum ether to yield 19.4-20.0 g (80-82%) of N-(tert-butoxycarbonyl)-β-iodoalanine methyl ester 3 as white to pale yellow crystals (Note 12).
C. N-(tert-Butoxycarbonyl)-β-[4-(methoxycarbonyl)phenyl]alanine methyl ester (4). A three-necked, 100-mL, round-bottomed flask equipped with an argon inlet adapter, reflux condenser, rubber septum, and magnetic stirbar (Note 13) is charged with 2.7 g (42 mmol) of zinc dust and 0.039 g (0.15 mmol) of I2. The flask is evacuated to 0.03 mm and heated with a heat gun for 10 min, flushed three times with argon, and then allowed to cool to room temperature. Dimethylformamide (1 mL) (Note 14) is added via syringe, and then a solution of 10.8 g (33 mmol) of iodide 3 in 14 mL of DMF is added dropwise via syringe to the well-stirred suspension of zinc dust. The reaction mixture is stirred for 30 min at 0°C to produce a solution of the zinc reagent and a suspension of excess zinc dust. The ice bath is removed and 7.9 g (30 mmol) of methyl 4-iodobenzoate (Note 15), 0.137 g (0.15 mmol) of tris(dibenzylideneacetone)dipalladium(0), and 0.183 g (0.6 mmol) of tri-o-tolylphosphine are added. The reaction mixture is then stirred at 60°C for 5 hr. The resulting mixture is poured into a conical flask containing 300 mL of water. An additional 100 mL of 10% citric acid solution is added in order to break the black emulsion. The aqueous layer is extracted with two 150-mL portions of diethyl ether, and the combined organic layers are washed with two 100-mL portions of water and 100 mL of brine. At this point, the organic extracts are decanted from a black solid [consisting of palladium and zinc(0), together with a bright white solid; (Note 16)], dried over magnesium sulfate, filtered, and concentrated by rotary evaporation to yield 9.1 g of a pale yellow solid. This material is purified by column chromatography (Note 17). The product is charged on a column of 200 g of silica gel (Sorbent 60A, 32-63 µm) and eluted with 1 L of 20% EtOAc-hexane. At that point, fraction collection (25-mL fractions) is begun, and elution is continued with 500 mL of 25% EtOAc-hexane and then 200 mL of 30% EtOAc-hexane. The desired product is collected in fractions 13-59 which are concentrated by rotary evaporation to yield 3.5-3.9 g (35-39%) of 4 as a white solid (Note 18).
2. Notes
1.
The checkers purchased
N-(tert-butoxycarbonyl)-L-serine methyl ester from Aldrich Chemical Company, Inc. The submitters prepared this compound, a thick amber oil, according to the procedure described in Dondoni, A.; Perrone, D.
Org. Synth. Coll. Vol. X,
2004, 320.
2.
The checkers purified
dichloromethane by pressure filtration through
activated alumina.
3.
The checkers purchased
4-DMAP and Me3N·HCl from Aldrich Chemical Company, Inc. Et3N was purchased from Alfa Aesar Chemical Company and was distilled from
CaH2. The submitters obtained
4-DMAP and Et3N from Lancaster and
Me3N·HCl from Aldrich Chemical Company, Inc.4.
TsCl was obtained from Aldrich Chemical Company, Inc. and purified by recrystallization according to the following procedure.
p-Toluenesulfonyl chloride (85 g) is dissolved in
150 mL of hot CHCl3 and
200 mL of petroleum ether (room temperature) is added in one portion to the clear, colorless solution. The resulting cloudy solution is clarified by addition of ca. 5 g of charcoal, stirred for 1 min, and filtered on a Büchner funnel. The filtrate is concentrated to ca. 1/5th of its original volume by rotary evaporation, and the solid which appears is collected by filtration and dried under reduced pressure (25°C, 0.03 mm) to afford
68 g of
TsCl as bright white crystals.
5.
Triethylamine addition must be carried out slowly to avoid base-promoted elimination of
p-toluenesulfonate in the final product.
6.
It may be necessary to scratch the flask in order to start crystallization.
7.
The product exhibits the following properties:
mp 74-76°C, lit.
7a 74-75°C;
[α]D20 +3.0 (methanol, c 2.0), lit.
7b +4.6 (methanol, c 2); Rf 0.24 (petroleum ether/EtOAc 3:1); IR (CH
2Cl
2) cm
−1: 3384, 2979, 1753, 1714, 1511, 1367, 1191, 1177;
1H NMR
pdf(500 MHz, CDCl
3) δ: 1.43 (s, 9 H), 2.46 (s, 3 H), 3.71 (s, 3 H), 4.29 (dd,
J = 10.1, 3.1 Hz, 1 H), 4.40 (dd,
J = 10.1, 3.1 Hz, 1 H), 4.50-4.53 (m, 1 H), 5.30 (d,
J = 7.9 Hz, NH), 7.37 (app d,
J = 7.9 Hz, 2 H), 7.77 (app d,
J = 8.2 Hz, 2 H);
13C NMR (125 MHz, CDCl
3) δ: 21.9, 28.4 (3 C), 53.1 (2 C), 69.7, 80.7, 128.2 (2 C), 130.1 (2 C), 132.5, 145.4, 155.1, 169.2; MS (EI)
m/z 314 (M+-CO
2Me, 20%), 300 [(M+-O
tBu), 8%], 258 [(M+-BocNH
2), 13%], 215 (11%), 155 (TolSO
2, 43%), 57 (100%); HRMS (EI)
m/z: [M - CO
2Me]+ calcd for C
14H
20NO
5S, 314.1057; found, 314.1059.
8.
A.C.S. Reagent grade acetone (99.5%) was purchased from Aldrich Chemical Company, Inc.9.
The checkers purchased
sodium iodide from Mallinckrodt Chemical Co. and dried it under vacuum (0.03 mm) at 100°C for 2 days.
10.
The reaction can be monitored by TLC: elution with
3:1 petroleum ether/ethyl acetate, R
f = 0.24 (
O-tosylserine), R
f = 0.60 (
iodoalanine).
11.
Due to the unstable nature of the
iodide, the solution should be cooled in a
ice-water bath at 0°C during concentration.
12.
Iodoalanine derivative 3 is also available from Aldrich Chemical Company, Inc. Compound
3 exhibits the following spectroscopic and physical properties:
mp 45-47°C, lit.
6b mp = 51°C;
[α]D20 −3.7 (MeOH), c 3.0, lit.
6b [α]D20 = −4.0 (c 3 methanol); R
f 0.60 (petroleum ether/AcOEt 3:1); IR (CH
2Cl
2) cm
−1: 1163, 1259, 1422, 1714, 1748, 2986, 3054, 3424;
1H NMR
pdf(500 MHz, CDCl
3) δ: 1.47 (s, 9 H), 3.55-3.67 (m, 2 H), 3.81 (s, 3 H), 4.53-4.54 (m, 1 H), 5.36 (d, J = 6.4 Hz, 1 H); C NMR (125 MHz, CDCl
3 δ: 8.6, 29.0 (3 C), 53.7, 54.4, 81.2, 155.5, 170.8,; MS (EI)
m/z 270 (M+-CO
2Me 25%), 214 [(M+-Boc), 8%], 57 (100%); HRMS (ESI)
m/z: [M + Na]+ calcd for C
9H
16INO
4, 352.0016; found, 352.0021; Anal. Calcd for C
9H
16INO
4: C, 32.8; H 4.9; N, 4.3. Found: C, 33.2; H 4.8; N, 4.1.
13.
The submitters used a
two-necked, round-bottomed flask equipped with a magnetic stirbar,
three-way stopcock connected to vacuum and a N
2 source, and a
solid addition tube. The addition tube was charged with
methyl 4-iodobenzoate,
tris(dibenzylideneacetone)dipalladium(0), and
tri-o-tolylphosphine. After the
zinc reagent was formed and the ice bath was removed, the solids contained in the addition tube were added in one portion by inverting the tube.
14.
DMF was distilled from
CaH2 and stored over activated 4Å molecular sieves.
15.
Methyl 4-iodobenzoate was purchased from Avocado Chemical Co. Zinc dust (< 10 microns, 95% purity) was purchased from Aldrich Chemical Company, Inc. The checkers purchased
Pd2dba3 and P(o-Tol)3 from Strem Chemicals, Inc. and
I2 from Mallinckrodt. The submitters purchased
Pd2dba3 and P(o-Tol)3 from Aldrich Chemical Company, Inc.16.
This solid was identified as
dimethyl biphenyl-4,4'-dicarboxylate:
1H NMR (200MHz, CDCl
3) δ: 3.96 (s, 6H), 7.67-7.72 (BB', 4H), 8.10-8.16 (AA', 4H);
mp 219-220°C, (lit.
mp 214-217°C: Catalogue handbook of Fine Chemicals. Aldrich 1999-2000).
17.
The submitters purified the product by the following procedure. The residual pale yellow solid is dissolved in
50 ml of diethyl ether and the remaining solid is filtered off
(Note 16). The filtrate is concentrated to a volume of ca. 25 mL, and the solution is allowed to crystallize at 0°C. Once crystallization begins,
50 mL of petroleum ether is added in two portions over 10 hr, and then crystallization is allowed to proceed overnight at 0°C. The white solid is collected by filtration and washed with a mixture of
3:1 petroleum ether-diethyl ether to afford
3.8 g of
4. Chromatographic purification of the mother liquor (5.5 × 18 cm of DSH silica gel 40-63 mm, elution with
1 L of petroleum ether/ethyl acetate 4:1 followed by
1.5 L of 3:1 petroleum ether-ethyl acetate) gives
2.5 g of
4 as a pale yellow solid. All the material is combined and recrystallized from diethyl
ether/petrol as above to yield
5.2 g (
47%) of
4 in two crops.
18.
The enantiomeric purity of compound
4 was higher than 99.5% as determined by HPLC (2-cm Supelco Si precolumn, 250 × 4.6 mm Chiralpak AS, 10%
isopropanol in heptane, 1mL/min, t
r = 14.5 min). The D enantiomer (t
r = 11.5 min) was not detected. Alternatively, the Boc protecting group of compound
4 (0.100 g) was removed by treatment with a solution prepared by addition of
100 µL of AcCl to
1.5 mL of methanol. Mosher's amides were formed using
i-Pr2NEt (1.1 equiv), EDCI (1.1 equiv), BtOH (1.1 equiv) and (R) or (S)-α-methoxy-α-(trifluoromethyl)phenylacetic acid (1.5 equiv), and 1H NMR (500 MHz) analysis of both amides showed ee > 98%. Compound
4 exhibits the following spectroscopic and analytical properties:
mp 82-83°C;
[α]D20 = −5.0 (acetone, c 1.0); IR (CH
2Cl
2) cm
−1: 3367, 2979, 2954, 1720, 1613, 1512, 1282, 1167; 1H NMR
pdf(500 MHz, CDCl
3) δ: 1.42 (s, 9 H), 3.10 (dd,
J = 13.7, 6.4 Hz, 1 H), 3.20 (dd,
J = 13.7, 5.8 Hz, 1 H), 3.91 (s, 3 H), 3.72 (s, 3 H), 4.61-4.63 (m, 1 H), 5.00 (d,
J = 7.9 Hz, NH), 7.21 (app d,
J = 7.9 Hz, 2 H), 7.98 (app d,
J = 8.5 Hz, 2 H);
13C NMR (125 MHz, CDCl
3) δ: 29.0 (3 C), 39.1, 52.8, 53.1, 54.9, 80.8, 129.6, 130.1 (2 C), 130.5 (2 C), 142.2, 155.7, 167.6, 172.7; MS (EI) m/z 337 (M+, 0.3%), 306 [(M+−OMe), 7%], 264 [(M+−O
tBu), 4%], 220 [(M+−BocNH
2), 83%], 150 (79%), 57 (100%); HRMS (ESI)
m/z: [M + Na]+ calcd for C
17H
23NO
6 360.1418; found, 360.1407; Anal. Calcd for C
17H
23NO
6: C, 60.51; H, 6.88; N, 4.15. Found: C, 60.3; H, 6.6; N, 4.0.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
The development of synthetic equivalents for the alanine β-anion is an important target for the synthesis of α-amino acids. A useful solution was provided by the discovery that organozinc reagents could be prepared from the iodoalanine derivative (
3) by insertion of activated
zinc into the
carbon-
iodine bond.
2 The resulting organometallic reagent could then be coupled with carbon electrophiles in the presence of a suitable
palladium catalyst. Furthermore, treatment of the organozinc reagent with copper cyanide gave the organocuprate, which could then couple directly with reactive electrophiles.
3The iodoalanine derivative is available in both enantiomeric forms and this method offers an extremely simple route to large numbers of non-natural amino acids.
4 Therefore a reliable and practical method for the synthesis of fully protected iodoalanine
3 in a multigram scale is highly desirable.
Although the tosylation reaction of fully protected serine has already been described,
5 we have found that extended reaction times are required when the reaction is carried out on 100-200 mmol scale, taking from 3 to 5 days to go to completion. As a result of longer reaction times, the amount of by-products is increased making the purification step more difficult.
Recently Tanabe and co-workers have found that several alcohols were smoothly and efficiently tosylated using tosyl chloride/
triethylamine and a catalytic amount of
trimethylamine hydrochloride as reagents.
6 Compared with the traditional method using
pyridine as solvent, this procedure has the merit of much higher reaction rates, and it avoids the side reaction in which the desired tosylate is converted into the corresponding chloride.
Stirring a mixture of tosylate and
NaI in
acetone at room temperature for four days produced the iodoalanine derivative
3 via a
pseudo-Finkenstein reaction. Although this reaction has been reported to proceed in shorter reaction times when carried out at reflux,
7 in our hands, an increase in the temperature of the reaction produced significant amounts of by-products and a poor mass balance. For instance, when the experimental procedure stated in step B was carried out at reflux of
acetone only 4 g of crude material was recovered. Three water-soluble products account for the rest of the mass balance, and one of them has been identified as
4-methoxycarbonyloxazolidin-2-one.
8 Better yields can be obtained when the reaction was carried out at reflux on a smaller scale (
2 g of compound
2) and under much more dilute conditions as reported.
7 It is our contention that due to the instability of the iodoalanine derivative, high temperatures during the reaction or the work-up promote decomposition of the product
via intramolecular nucleophilic attack by the carbamate moiety. Nevertheless, compound
3 could be kept in the refrigerator for one year without evidence of decomposition.
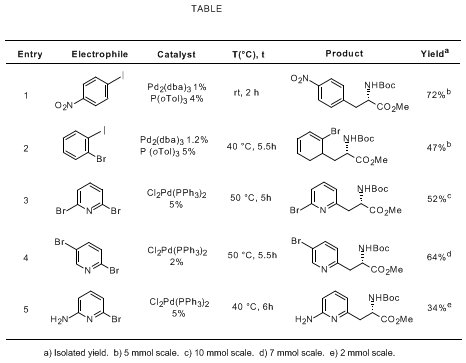
The procedure in Section C is representative of the synthesis of non-natural α-amino acids featuring the palladium cross coupling reaction of a β-alanine organozinc derivative with aromatic electrophiles. This methodology has been successfully extended with modifications to both the electrophile and the catalyst as shown in the Table.
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
N-(tert-Butoxycarbonyl)-L-serine methyl ester:
L-Serine, N-[(1,1- dimethylethoxy)carbonyl]-, methyl ester; (2766-43-0)
N-(tert-Butoxycarbonyl)-β-iodoalanine methyl ester:
L-Alanine, N-[(1,1- dimethylethoxy)carbonyl]-3-iodo-, methyl ester; (93267-04-0)
N-(tert-Butoxycarbonyl)-β-4-(methoxycarbonyl)phenyl]alanine methyl ester:
L-Phenylalanine, N-[(1,1-dimethylethoxy)carbonyl]-4-(methoxycarbonyl)-, methyl ester; (160168-19-4)
4-Dimethylaminopyridine:
4-Pyridinamine, N,N-dimethyl-; (1122-58-3)
Trimethylamine hydrochloride:
Methanamine, N,N-dimethyl-, hydrochloride; (593-81-7)
p-Toluenesulfonyl chloride:
Benzenesulfonyl chloride, 4-methyl-; (98-59-9)
Triethylamine:
Ethanamine, N,N-diethyl-; (1221-44-8)
Sodium iodide:
Sodium iodide (NaI); (7681-82-5)
Sodium thiosulfate:
Thiosulfuric acid (H2S2O3), disodium salt; (7772-98-7)
Methyl 4-iodobenzoate:
Benzoic acid, 4-iodo-, methyl ester; (619-44-3)
Tris(dibenzylideneacetone)dipalladium:
tris[μ-[(1,2-η:4,5-η)-(1E,4E)-1,5-diphenyl-1,4- pentadien-3-one]]di-; (51364-51-3)
Tri-o-tolylphosphine:
Phosphine, tris(2-methylphenyl)-; (6163-58-2)
Zinc; (7440-66-6)
Iodine; (7553-56-2)
Dimethyl biphenyl-4,4'-dicarboxylate:
[1,1'-Biphenyl]-4,4'- dicarboxylic acid, dimethyl ester; (792-74-5)
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved