Org. Synth. 2005, 81, 225
DOI: 10.15227/orgsyn.081.0225
SYNTHESIS AND USE OF GLYCOSYL PHOSPHATES AS GLYCOSYL DONORS
Submitted by Kerry R. Love and Peter H. Seeberger
1.
Checked by Youseung Shin and Dennis P. Curran.
1. Procedure
A. Dibutyl 3,4,6-tri-O-benzyl-2-O-pivaloyl-D-glucopyranosyl phosphate. A dry 500-mL, round-bottomed flask is charged with 5.00 g (12.0 mmol) of 3,4,6-tri-O-benzyl-D-glucal (Note 1) and 7 mL of toluene (Note 2). The toluene is removed azeotropically on a rotary evaporator and this process is repeated two more times. The residue is dried under vacuum for 30 min. Under a flow of nitrogen, the flask is charged with a magnetic stirbar and 50 mL of dichloromethane (Note 2) and the resulting solution is cooled to 0°C. A solution of dimethyldioxirane (200 mL of a 0.08M solution, 16.0 mmol) (Note 3) is added to the flask via cannula and the reaction mixture is stirred at 0°C for 10 min (Note 4). Volatile materials are then removed by rotary evaporation at 0°C (Note 5) and the resulting white residue is dried at 2 mm for 5 min. The flask is equipped with a rubber septum fitted with a nitrogen inlet needle and the residue is dissolved in 40 mL of CH2Cl2 and the solution is cooled to −78°C. A solution of dibutyl phosphate (2.85 mL, 14.4 mmol) (Note 6) in 10 mL of dichloromethane is added via cannula. After 10 min at −78°C, the reaction mixture is warmed to 0°C and 4-(dimethylamino)pyridine (DMAP) (5.86 g, 48.0 mmol) and pivaloyl chloride (2.96 mL, 24.0 mmol) are added (Note 7). The solution is stirred for 30 min at 0°C, then allowed to warm to room temperature and stirred for 16 hr (Note 8). A solution of 25% ethyl acetate in hexanes is added and the resulting suspension is filtered through a pad of silica gel. The filtrate is concentrated to a clear oil and purified by column chromatography through a short plug of 80 g of silica gel (elution with 35% ethyl acetate-hexanes) (Note 9) to afford 5.86 g (67%) of dibutyl 3,4,6-tri-O-benzyl-2-O-pivaloyl-D-glucopyranosyl phosphate as a colorless oil (Notes 10, 11).
B. 3,4,6-Tri-O-benzyl-2-O-pivaloyl-β-D-glucopyranosyl-(1→6)-1,2:3,4-di-O-isopropylidene-α-D-galactopyranoside. A dry 500-mL, round-bottomed flask is charged with dibutyl 3,4,6-tri-O-benzyl-2-O-pivaloyl-D-glucopyranosyl phosphate (5.89 g, 8.11 mmol), 1,2:3,4-di-O-isopropylidene-α-D-galactopyranose (1.92 g, 7.37 mmol) (Note 12), and 7 mL of toluene (Note 2). The toluene is removed azeotropically on a rotary evaporator and this process is repeated two more times. The residue is dried under vacuum for 1 hr. The flask is equipped with a rubber septum fitted with a nitrogen inlet needle and a magnetic stirbar, and 100 mL of CH2Cl2 is added. The resulting solution is cooled to −60°C. Trimethylsilyl triflate (TMSOTf) (1.47 mL, 8.11 mmol) (Note 13) is added and the solution is allowed to warm slowly to −40°C (Note 14). After 30 min, the reaction is quenched by addition of 15 mL of triethylamine (Note 15) and allowed to warm to room temperature. The reaction mixture is concentrated by rotary evaporation and the residue is purified by column chromatography on 140 g of silica gel (elution with 30% ethyl acetate-hexanes) to afford 5.09 g (85%) of 3,4,6-tri-O-benzyl-2-O-pivaloyl-D-glucopyranosyl-(1→6)-1,2:3,4-di-O-isopropylidene-α-D-galactopyranoside as a colorless oil (Notes 16, 17).
2. Notes
1.
Tri-O-benzyl-D-glucal (97%) was purchased from Aldrich Chemical Company, Inc. and was used as supplied without further purification.
2.
Toluene and dichloromethane (CH2Cl2) were purified by a JT Baker Cycle-Tainer Solvent Delivery System.
3.
Dimethyldioxirane was prepared according to the literature procedure
2 as an 0.08M solution and was dried over 4å beaded molecular sieves for 24 hr prior to use.
4.
Analytical thin-layer chromatography was performed on E. Merck silica gel 60 F
254 plates (0.25 mm) and compounds were visualized by dipping the plates in a cerium sulfate-ammonium molybdate solution followed by heating.
5.
Expedient removal of the volatiles after the epoxidation with
dimethyldioxirane is crucial to achieve reproducible yields because the epoxide is extremely water sensitive.
6.
Dibutyl phosphate (97%) was purchased from Fluka and was used as supplied without further purification.
7.
DMAP (99%) and pivaloyl chloride (99%) were purchased from Aldrich Chemical Company, Inc. and were used as supplied without further purification.
8.
The submitters recommend that the reaction be worked up immediately upon reaching room temperature, but the checkers found that it was not detrimental to allow the reaction to stir overnight at room temperature prior to workup.
9.
Prolonged chromatography when purifying glycosyl phosphates will lead to hydrolysis and decomposition of the product.
10.
Early fractions were mixtures of α/β anomers while later fractions contained pure β-anomer. The fractions were combined to give the product as a 9:1 mixture of β/α-anomers. Analytical data for this compound are as follows: (β-phosphate) R
f 0.29 (
ethyl acetate:hexane, 1:3);
[α]D24 −1.9 (c = 1.50, CH2Cl2); IR (thin film) cm
−1: 2946, 1740, 1454, 1282, 1016;
1H NMR
pdf (500 MHz, CDCl
3) δ: 0.96-0.88 (m, 6H), 1.20 (s, 9H), 1.40-1.34 (m, 4H), 1.64-1.59 (m, 4H), 3.64-3.61 (m, 1H), 3.78-3.70 (m, 3H), 3.82 (t,
J = 9.5 Hz, 1H), 4.08-4.00 (m, 4H), 4.51 (d,
J = 11.0 Hz, 1H), 4.69-4.54 (m, 2H), 4.70 (d,
J = 11.0 Hz, 1H), 4.80-4.75 (m, 2H), 5.17 (app t,
J = 8.5 Hz, 1H), 5.24 (app t,
J = 7.3 Hz, 1H), 7.16-7.14 (m, 2H), 7.33-7.25 (m, 13H);
13C NMR (125 MHz, CDCl
3) δ: 14.0, 19.1, 26.9, 32.7, 39.2, 68.1, 68.2, 68.4, 73.3, 73.9, 75.9, 76.2, 83.1, 97.0, 127.6, 128.0, 128.1, 128.2, 128.3, 128.7, 138.1, 138.2, 177.2, (d,
JC-P = 5.0 Hz);
31P NMR (200 MHz, CDCl
3) δ: −2.2; FAB MS
m/z (M)
+ calcd 726.3532, obsd 726.3537. (α-Phosphate) R
f 0.36 (
ethyl acetate:hexane, 1:3);
[α]D24 +50.5 (c = 0.63, CH2Cl2); IR (thin film) cm
−1: 2960, 2872, 1736, 1454, 1282;
1H NMR
pdf (500 MHz, CDCl
3) δ: 0.97-0.91 (m, 6H), 1.24 (s, 9H), 1.44-1.36 (m, 4H), 1.86-1.61 (m, 4H), 3.68 (d,
J = 11.0 Hz, 1H), 3.86-3.79 (m, 2H), 4.10-4.02 (m, 5H), 4.56-4.50 (m, 3H), 4.63 (d,
J = 11.5 Hz, 1H), 4.83-4.80 (m, 3H), 4.99-4.97 (m, 1H), 5.85 (dd,
J = 1.8, 6.4 Hz, 1H), 7.18-7.15 (m, 2H), 7.35-7.27 (m, 13H);
13C NMR (125 MHz, CDCl
3) δ: 13.8, 18.8, 27.3, 32.4, 32.5, 39.0, 67.8, 67.9, 68.0, 68.2, 72.6, 72.7, 73.7, 75.4, 75.6, 79.5, 94.7, 127.6, 127.7, 127.9, 128.0, 128.1, 128.2, 128.3, 128.5, 128.6, 138.0, 138.1, 138.3, 177.7, (d,
JC-P = 5.5 Hz);
31P NMR (200 MHz, CDCl
3) δ: −2.5; FAB MS
m/z (M)
+ calcd 726.3532, obsd 726.3537.
11.
Practical considerations limit the scalability of this reaction due to the highly reactive and water sensitive intermediates formed. Furthermore, the time required for removal of large amounts of solvent
in vacuo allows for the opening of the intermediate epoxide leading to diol formation.
12.
1,2:3,4-Di-O-isopropylidene-α-D-galactopyranoside (97%) was purchased from Aldrich Chemical Company, Inc. and was used as supplied without further purification.
13.
TMSOTf (99%) was purchased from Acros Organics and was used as supplied without further purification.
14.
The submitters report that the less reactive α-phosphate can be completely consumed by allowing the reaction mixture to warm to −20°C prior to workup.
15.
Triethylamine (98%) was purchased from J.T.Baker and was used as supplied without further purification.
16.
The product was obtained in
90-91% yield in other runs. Analytical data for this compound are as follows:
[α]D24−45.2 (c = 2.34, CH2Cl2); IR (thin film) cm
−1: 3029, 2978, 2933, 2904, 1741, 1134, 1028;
1H NMR
pdf (500 MHz, CDCl
3) δ: 1.21 (s, 9H), 1.31 (s, 3H), 1.32 (s,3H), 1.43 (s, 3H), 1.51 (s, 3H), 3.53-3.50 (m, 1H), 3.63-3.59 (m,1H), 3.76-3.69 (m, 4H), 3.97-3.94 (m, 1H), 4.10-4.07 (m, 1H), 4.29-4.25 (m, 2H), 4.46 (d,
J = 8.0 Hz, 1H), 4.58-4.53 (m, 3H), 4.64 (d,
J = 8.0 Hz, 1H), 4.79-4.69 (m, 3H), 5.10 (app t,
J = 8.5 Hz), 5.49 (d,
J = 5.0 Hz, 1H), 7.19-7.15 (m, 2H), 7.39-7.24 (m, 13H);
13C NMR (125 MHz, CDCl
3) δ: 24.7, 25.4, 26.3, 26.4, 27.5, 39.1, 67.4, 69.0, 70.9, 71.5, 73.3, 73.9, 75.2, 75.7, 78.1, 83.6, 96.6, 101.8, 108.8, 109.5, 127.7, 127.9, 127.8, 128.0, 128.1, 128.2, 128.6, 128.7, 138.4, 138.5, 177.1; FAB MS
m/z (M)
+ calcd 776.3772, obsd 776.3770.
17.
Continued elution gave unreacted α-phosphate (
0.125 g,
2%).
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
The role of complex carbohydrates in biological processes is now widely appreciated. Oligosaccharides and glycoconjugates are essential for many cellular events, such as recognition, adhesion and signaling between cells.
3 Carbohydrates have also been implicated in a variety of disease states including cancers
4 and in a host of bacterial and viral infections.
5 The widespread biological implications of carbohydrates have rendered them targets of intense study. Microheterogeneity of naturally derived oligosaccharides, however, limits the access to pure materials in appreciable quantity. Chemical synthesis, therefore, remains the best way procure material for biological investigations.
Advances in carbohydrate chemistry, particularly in the development of powerful glycosylating agents, have provided access to molecules of biological interest. In particular, glycosyl trichloroacetimidates,
6 glycosyl fluorides,
7 thioglycosides
8 and n-pentenyl glycosides
9 have each been used in the construction of complex oligosaccharides. Limitations of these glycosyl donors, including lengthy syntheses, difficult activation conditions, and long reaction times, have lead us to develop glycosyl phosphates for the facile construction of a variety of glycosidic linkages.
Glycosyl phosphates can be synthesized from a variety of intermediates; anomeric lactols
10,11 and 1,2-anhydrosugars
12 are most typically used. Additionally, other glycosyl donors, such as glycosyl trichloroacetimidates
13 and
n-pentenyl glycosides,
14 may be converted into glycosyl phosphates. The aforementioned procedure generates the 1,2-anhydrosugar from a suitably protected glycal with dimethyldioxirane. Opening of the anhydrosugar at low temperature using a phosphate diester, followed by acylation of the C
2-hydroxyl group leads to fully protected glycosyl phosphates (Figure 1).
12This method is attractive for three reasons: First, glycal precursors are desirable starting materials because they require the differentiation of only three hydroxyl groups, each having unique reactivity. Generation of phosphates from anomeric lactols or other glycosyl donors requires many synthetic steps for the differential protection of the five hydroxyl groups present. Second, the procedure can be carried out in one pot providing quick access to the desired glycosyl phosphates in high yields. Finally, choice of solvent for the opening of the anhydrosugar can lead to either alpha- or beta-enriched phosphates.
12Initial work by Ikegami and coworkers showed that glycosyl phosphates are highly reactive glycosylating agents.
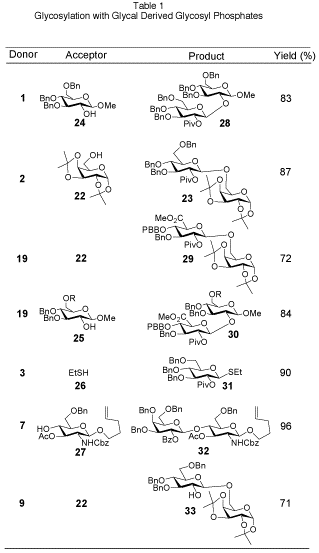
15 Activation of the glycosyl phosphate by addition of a stoichiometric amount of a Lewis acid in the presence of an acceptor quickly forms the desired disaccharide in good yield (Table 1). Glycosyl phosphates have been successfully employed in the synthesis of both
O- and
C-glycosides,
16 including biologically relevant compounds such as the Leishmania antigenic tetrasaccharide
17 and the H-type II blood group pentasaccharide.
18 Use of phosphates on solid-support in an automated fashion further demonstrates their utility as glycosyl donors.
19Figure 1
Glycosyl Phosphates Prepared from Glycals

Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
Dibutyl 3,4,6-tri-O-benzyl-2-O-pivaloyl-D-glucopyranosyl phosphate:
β-D- Glucopyranose, 3,4,5-tris-O-(phenylmethyl)-, 1-(dibutyl phosphate) 2-(2,2- dimethylpropanoate); (223919-63-7)
3,4,6-Tri-O-benzyl-D-glucal:
D-arabino-Hex-1-enitol, 1,5-anhydro-2-deoxy-3,4,6-tris-O- (phenylmethyl)-; (55628-54-1)
Dibutyl phosphate:
Phosphoric acid, dibutyl ester; (107-66-4)
4-(Dimethylamino)pyridine:
4-Pyridinamine, N,N-dimethyl-; (1122-58-3)
Pivaloyl chloride:
Propanoyl chloride, 2,2-dimethyl-; (3282-30-2)
3,4,6-Tri-O-benzyl-2-O-pivaloyl-β-D-glucopyranosyl-(1→6)-1,2:3,4-di-O-isopropylidene- α-D-galactopyranoside:
α-D-Galactopyranose, 6-O-[2-O-(2,2-dimethyl-1- oxopropyl)-3,4,6-tris-O-(phenylmethyl)-β-D-glucopyranosyl]-1,2:3,4-bis-O-(1-methylethylidene)-; (219122-26-6)
1,2:3,4-Di-O-isopropylidene-α-D-galactopyranose:
α-D-galactopyranose, 1,2:3,4-bis-O- (1-methylethylidene)-; (4064-06-6)
Trimethylsilyl triflate:
Methanesulfonic acid, trifluoro-, trimethylsilyl ester; (27607-77-8)
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved