Org. Synth. 2005, 82, 126
DOI: 10.15227/orgsyn.082.0126
IRIDIUM-CATALYZED C-H BORYLATION OF ARENES AND HETEROARENES: 1-CHLORO-3-IODO-5-(4,4,5,5-TETRAMETHYL-1,3,2-DIOXABOROLAN-2-YL)BENZENE AND 2-(4,4,5,5,-TETRAMETHYL-1,3,2-DIOXABOROLAN-2-YL)INDOLE
[(2-(3-Chloro-5-iodophenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane) (2-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indole)]
Submitted by Tatsuo Ishiyama, Jun Takagi, Yusuke Nobuta, and Norio Miyaura
1.
Checked by Yili Shi, Daniel J. Weix, and Jonathan A. Ellman.
1. Procedure
Caution! The reactions produce hydrogen gas and should be conducted in a well-ventilated hood.
A. 1-Chloro-3-iodo-5-(4,4,5,5-tetramethyl-1,3,2,-dioxaborolan-2-yl)benzene. A 50-mL, two-necked, round-bottomed flask is fitted with a magnetic stirring bar, a rubber septum, and a condenser to which a nitrogen inlet and an oil bubbler are attached, and flushed with nitrogen (Note 1). The septum is removed and the flask is charged with bis(η4-1,5-cyclooctadiene)-di-μ-methoxy-diiridium(l) ([Ir(OMe)(COD)]2) (33 mg, 0.050 mmol) (Note 2), 4,4'-di-tert-butyl-2,2'-bipyridine (dtbpy) (27 mg, 0.10 mmol) (Note 3), and bis(pinacolato)diboron (2.67 g, 10.5 mmol) (Note 4). The septum is again placed on the flask, and the flask is purged with nitrogen for 1 min. Hexane (30 mL) (Note 5) is added by syringe, and the flask is immersed in an oil bath that is maintained at 50 °C. The mixture is stirred for 10 min to give a dark red solution. The flask is charged with 1-chloro-3-iodobenzene (4.75 g, 19.9 mmol) by syringe (Note 6), and then the resulting dark red solution is stirred at 50 °C for 6 hr (Note 7). The mixture is removed from the oil bath, allowed to cool to room temperature, and poured into a separatory funnel. The reaction flask is rinsed with hexane (2 × 10 mL). The rinses and water (30 mL) are added to the separatory funnel, the funnel is shaken, the layers are separated, and the organic extracts are dried over anhydrous magnesium sulfate. The drying agent is removed by filtration and is washed with hexane (3 × 10 mL), and the filtrate is concentrated on a rotary evaporator to give a dark brown oil. The oil is distilled under reduced pressure (Note 8) to afford 6.11-6.20 g (84-86%) of 1-chloro-3-iodo-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzene as a white solid, mp 58.6-60.7 °C (Note 9).
B. 2-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)indole. A 50-mL, two-necked, round-bottomed flask is fitted with a magnetic stirring bar, a rubber septum, and a condenser to which a nitrogen inlet and an oil bubbler are attached, and flushed with nitrogen (Note 1). The septum is removed and the flask is charged with bis(η4-1,5-cyclooctadiene)-di-μ-methoxy-diiridium(l) ([Ir(OMe)(COD)]2) (50 mg, 0.075 mmol) (Note 2) and 4,4'-di-tert-butyl-2,2'-bipyridine (dtbpy) (40 mg, 0.15 mmol) (Note 3). The septum is again placed on the flask, and the flask is purged with nitrogen for 1 min. Hexane (30 mL) (Note 5) and pinacolborane (4.79 mL, 33.0 mmol) (Note 10) are added by syringe, and the flask is immersed in an oil bath that is maintained at 25 °C. The mixture is stirred for 10 min. to give a dark red solution. The flask is charged with indole (3.51 g, 30.0 mmol) (Note 11), and then the resulting dark red suspension is stirred at 25 °C for 4 hr. The mixture is removed from the oil bath, allowed to cool to room temperature, and poured into a separatory funnel. The reaction flask is rinsed with ether (2 × 20 mL). The rinses and water (30 mL) are added to the separatory funnel, the funnel is shaken, the layers are separated, and the organic extracts are dried over anhydrous magnesium sulfate. The drying agent is removed by filtration and is washed with ether (3 × 10 mL), and the filtrate is concentrated on a rotary evaporator to give a dark brown oil. The oil is distilled under reduced pressure (Note 12) to afford 5.33-5.40 g (73-74%) of 2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)indole as a colorless viscous oil, bp 149-153 °C (0.15 mm) (Note 13).
2. Notes
1.
All glassware is pre-dried in an oven at 120 °C for 1 hr, assembled while hot, and allowed to cool under a stream of
nitrogen.
2.
[Ir(OMe)(COD)]2 is prepared from
[IrCl(COD)]2 by the reported procedure.
2 The complex can be handled in air, and stored in a tightly closed bottle and in a freezer. Checkers stored the complex in a vial in a nitrogen-filled glove-bag at rt.
3.
4,4'-Di-tert-butyl-2,2'-bipyridine (98%) was purchased from Aldrich Chemical Company, Inc., and used without further purification.
4.
Bis(pinacolato)diboron is prepared according to the literature procedure,
3 and dried under reduced pressure (0.1 mm) at room temperature for 16 hr. prior to use. The reagent is air-stable and commercially available. The checkers purchased
bis(pinacolato)diboron (98%) from Aldrich and used it without further purification.
5.
Hexane is distilled from
lithium aluminum hydride under nitrogen before use. The checkers purified hexanes by passage through a column of activated
alumina (type A2, size 12 × 32, Purifry Co.) followed by nitrogen sparge. The hexanes were stored over MS 3Å in a nitrogen-filled glove-box.
6.
1-Chloro-3-iodobenzene was purchased from Tokyo Kasei Kogyo Co., Ltd., and distilled from molecular sieves 4Å under nitrogen before use. The checkers purchased
1-chloro-3-iodobenzene (98%) from Aldrich.7.
The reaction using 1.5 mol% of the catalyst is completed within 4 hr at room temperature, but the decreasing the amount of catalyst to 0.5 mol% sufficiently slows the rate of the reaction that heating to 50 °C is required to complete the reaction in a reasonable amount of time.
8.
Bp 145-148 °C (0.3 mm)
1-Chloro-3-iodobenzene (28 mg) that is unreacted or is generated by protodeboration of the product is also obtained, bp 60 °C (0.05 mm). The checkers found that either simple distillation with a short-path distillation head or Kugelrohr distillation can be used. To avoid contamination of the product with lower boiling materials (eg starting materials or pinacol), the mixture should be heated until
1-chloro-3-iodobenzene and
pinacol have distilled. The apparatus is then allowed to cool, is disassembled, and is cleaned, before the product is distilled over.
9.
Gas chromatographic analysis of the product (Hitachi G-3500, OV-101 on UniportB, a glass column, 3 mm × 2 m) shows that the chemical purity is 97-99%. The spectral data are as follows:
1H NMR
pdf (400 MHz, CDCl
3) δ 1.30 (s, 12H), 7.72 (d, 1H,
J = 2.0), 7.78 (t, 1H,
J = 1.8), 7.99 (t, 1H,
J = 0.8);
13C NMR (100 MHz, CDCl
3) δ 24.8, 84.5, 94.2, 133.7, 134.7, 139.4, 141.4;
11B NMR (128.3 MHz, CDCl
3) δ 29.58 (BF
3·OEt
2 as external reference, δ 0.00); HRMS calcd for C
12H
15BCllO
2[M
+], 363.9899, found 363.9890.
10.
Pinacolborane (97%) was purchased from Aldrich Chemical Company, Inc., and used without further purification.
Pinacolborane is very sensitive to moisture and was troublesome to store for any period of time. The checkers found it could be stored under nitrogen at −30 °C in a sealable tube.
11.
Indole was purchased from Tokyo Kasei Kogyo Co., Ltd., and dried under reduced pressure (0.1 mm) at room temperature for 16 hr prior to use. The checkers purchased
indole (99%+) from Aldrich.
12.
The checkers accomplished the distillation with the apparatus below.
It was found that distillation with a traditional short-path led to clogging of the apparatus by both remaining indole starting material and the highly viscous product.
Indole (29 mg) that is unreacted or resulted by protodeboration of the desired product is also obtained, bp 120° C (0.7 mm). To avoid contamination of the product with lower boiling materials (eg starting materials or pinacol), the mixture should be heated until
indole and
pinacol have distilled over. The apparatus is then allowed to cool, is disassembled, and is cleaned, before the product is distilled over.
13.
Gas chromatographic analysis of the product (Hitachi G-3500, OV-101 on Uniport B, a glass column, 3 mm × 2 m) shows that the chemical purity is 96-99%. The spectral data are as follows:
1H NMR
pdf (400 MHz, CDCl
3) δ 1.37 (s, 12H), 7.10 (dt, 1H,
J = 0.8, 8.7), 7.12 (d, H,
J = 0.7), 7.23 (dt, 1H,
J = 1.1, 7.6), 7.39 (dd, 1H,
J = 1.0, 8.3), 7.68 (dd, 1H,
J = 1.0, 8.1), 8.56 (br s, 1H);
13C NMR (100 MHz, CDCl
3) δ 24.8, 84.1, 111.2, 113.8, 119.8, 121.6, 123.6, 128.3, 138.2;
11B NMR (128.3 MHz, CDCl
3) δ 28.45 (BF
3·OEt
2 as external reference, δ 0.00); HRMS calcd for C
14H
18BNO
2[M
+], 243.1431, found 243.1423.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Aryl- and heteroarylboron derivatives are important class of compounds that have been applied to various fields of chemistry.
4 Traditional methods for their synthesis are based on the reactions of trialkylborates with aryl- and heteroarylmagnesium or lithium reagents.
5 Pd-catalyzed cross-coupling of aryl and heteroaryl halides with tetra(alkoxo)diborons
6 or di(alkoxo)boranes
7 is a milder variant where the preparation of
magnesium and
lithium reagents is avoided.
Alternatively, the catalytic C-H borylation of arenes and heteroarenes, first reported by Smith,
8 is highly attractive as a halide-free process for the synthesis of aryl- and heteroarylboron compounds.
9 Among the catalysts developed to date, the combination of 1/2[Ir(OMe)(COD)]
2 and dtbpy described here exhibits high activity, which allows the formation of aryl- and heteroarylboronates in high yields at room temperature from an equimolar equivalent of
bis(pinacolato)diboron (pin
2B
2, pin = Me
4C
2O
2) or
pinacolborane (pinBH) and arenes or heteroarenes (Table 1).
9j-lThe regiochemistry of arene borylation is primarily controlled by the steric effects of substituents. The reaction occurs at C-H bonds located meta or para to a substituent in preference to those located ortho. Thus, 1,2- and 1,4-disubstituted arenes bearing identical substituents yield arylboronates as single isomers. The borylation of 1,3-disubstituted arenes proceeds at the common meta position; therefore, regioisomerically pure products are obtained even for two distinct substituents on the arenes. In the case of five-membered heteroarenes, the electronegative heteroatom causes the C-H bonds at the α-positions to be active so that the borylation occurs at the α-positions. Thus, the regioselective monoborylation of 2-substituted or benzo-fused substrates can be possible. Although a mixture of 2-borylated and 2,5-diborylated products is formed from unsubstituted substrates, both products are selectively obtained by reactions with the appropriate ratio of substrate and reagent. On the other hand, six-membered heteroarenes such as pyridine shows significantly lower reactivity due to strong coordinating ability of the basic nitrogen for the catalyst. Exceptionally, 2,6-disubstituted pyridines undergo smooth borylation at the 4-position.
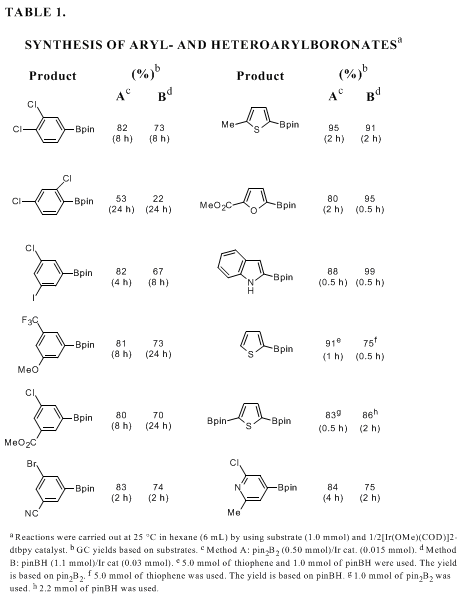
Functional group tolerance of the borylation is quite high. The reaction selectively occurs at the C-H bond for substrates possessing Cl, Br, I, CF3, OMe, CO2Me, and CN groups. The reaction occurs only at the aromatic C-H bonds even when the substrate has weaker benzylic C-H bonds.
Aryl- and heteroarylboronic acids and esters have been used for the synthesis of biaryls via the
palladium-catalyzed cross-coupling reaction with aryl electrophiles.
10 Sequential reactions involving aromatic C-H borylation and cross-coupling with aryl electrophiles in the same flask provide an efficient and convenient route to unsymmetrical biaryls (eq. 1).
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
Bis(η4-1,5-cyclooctadiene)-di-μ-methoxy-diiridium(l) ([Ir(OMe)(COD)]2):
Bis[(1,2,5,6-η)-1,5-cyclooctadiene]di-μ-methoxydiiridium; (12148-71-9)
4,4'-Di-tert-butyl-2,2'-bipyridine (dtbpy):
4,4'-Bis(1,1-dimethylethyl)-2,2'-bipyridine: (72914-19-3)
Bis(pinacolato)diboron: 4,4,4',4',5,5,5',5'-octamethyl-2,2'-Bi-1,3,2-dioxaborolane; (73183-34-3)
1-Chloro-3-iodobenzene; (625-99-0)
Pinacolborane:
4,4,5,5-Tetramethyl-1,3,2-dioxaborolane; (25015-63-8)
Indole:
1H-Indole; (120-72-9)
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved