Org. Synth. 2006, 83, 200
DOI: 10.15227/orgsyn.083.0200
2-SUBSTITUTED-1,3-CYCLOHEXADIENES BY INTERMOLECULAR, METHYLENE-FREE TANDEM ENYNE METATHESIS
[2-(Benzoyloxymethyl)-1,3-cyclohexadiene]
Submitted by Amol A. Kulkarni and Steven T. Diver
1.
Checked by Masakatsu Shibasaki and Keisuke Maki.
1. Procedure
A. Propargyl Benzoate. Into an oven-dried, argon-cooled 1-L round-bottomed flask equipped with magnetic stirbar and rubber septum is added CH2Cl2 (250 mL) (Note 1), which is maintained under an atmosphere of argon (Note 2). Propargyl alcohol (13.6 mL, 240 mmol, 1.2 equiv) and triethylamine (Note 3) (42 mL, 300 mmol, 1.5 equiv) are added by gas-tight syringe, followed by the addition of DMAP (Note 4) (2.4 g, 20 mmol, 0.1 equiv). The reaction mixture is subsequently cooled in an ice bath for 30 min. Benzoyl chloride (Note 5) (23.2 mL, 200 mmol, 1.0 equiv) is then added slowly by syringe over 15 min. The reaction mixture is stirred in the ice bath for an additional 30 min and then warmed to room temperature and stirred for 4 h. The reaction is quenched by addition of ice cold water (300 mL). The contents are transferred into a 1-L separatory funnel. The organic layer is washed with 1 M HCl (2 × 300 mL), followed by distilled water (300 mL) and brine (300 mL). The organic layer is separated, dried (Na2SO4) and concentrated under vacuum (rotary evaporator, 8–9 mmHg, 24 °C) (Note 6). The crude yellow oil is purified by bulb-to-bulb distillation (80 °C pot temperature) under reduced pressure (1–2 mmHg) to yield the desired ester (28.4–28.9 g, 89–90% yield) as a colorless oil (Note 7).
B. 2-(Benzoyloxymethyl)-1,3-Cyclohexadiene. Into an oven-dried, argon-cooled
100-mL Schlenk tube (Note 8) equipped with a magnetic stirbar, rubber septum and a cold finger condenser
(Note 9) under an atmosphere of
argon (Note 2) is added all-
cis polybutadiene (8.66 g, 160 mmol, 8 equiv) (Note 10) followed by
40 mL CH2Cl2 (Note 1). To the Schlenk tube is added Grubbs second generation
catalyst (Note 11) (271 mg, 0.32 mmol, 1.6 mol %) and the flask is then immersed in an oil bath maintained at 45 °C.
Propargyl benzoate (3.2 g, 20 mmole) is dissolved in about
1 mL of CH2Cl2 and this solution is added to the suspension of
polybutadiene in
CH2Cl2 over a period of 2 h by means of syringe pump addition with a gas-tight syringe connected to a short section of teflon tubing
(Note 12). The reaction flask is maintained at 45 °C during the addition. After the addition is complete, the syringe is rinsed with about
1 mL of CH2Cl2 and the contents are added to the reaction mixture over about 10 min. The reaction mixture is stirred at 45 °C for another 30 min. At this stage,
ethyl vinyl ether (Note 13) (612 µL, 0.64 mmol, 2 equiv based on ruthenium catalyst) is added and heating is continued for an additional 30 min. The reaction mixture is concentrated under vacuum (rotary evaporator, 8–9 mmHg, 23–25 °C) to give a brown oil which is then partially purified by passing through a plug of
silica gel (50 g; 2 × 10 cm) (Note 14) eluting with
0.8 % ethyl acetate in hexanes. The first 250 mL of the eluent is discarded
(Note 15). Then
700 mL of 4% ethyl acetate in hexanes is used to elute the desired
cyclohexadiene along with other oligomeric materials. The eluate is concentrated under vacuum (rotary evaporator, 10 mmHg, 25–40 °C). The viscous yellow oil thus obtained is further purified by means of bulb-to-bulb distillation (checker; 120–130 °C pot temperature/submitter; 124–128 °C) at reduced pressure (1–2 mmHg) to afford the desired
cyclohexadiene derivative as a colorless oil (
2.64–2.69 g,
62–63 % yield) (Notes
16,
17,
18). The
1H and
13C NMR spectra of the isolated compound match previously reported characterization data.
2
2. Notes
1.
The submitters used argon-sparged
dichloromethane (CH2Cl2, Mallinckrodt, 99%) drawn from a solvent purifier (two 0.05 m × 1 m stainless steel columns packed with activated alumina) maintained under argon immediately prior to use. The checkers used anhydrous
dichloromethane, which was used as received from Kanto Chemical Co. No degassing of the solvent was performed.
2.
Argon was supplied statically through a gas manifold with a mineral oil bubbler to generate slight back-pressure.
3.
The submitters used
triethylamine (Acros, 99%) distilled from CaH
2. The checkers used
triethylamine (Aldrich, 99.5%) as received.
4.
DMAP (99%) was used as received from Aldrich Chemical Co.5.
The submitters used
benzoyl chloride (Baker, 99.8%) as received. The checkers used
benzoyl chloride (Aldrich, 99%) as received.
6.
The submitters used a diaphragm-type vacuum pump equipped with a primary dry ice-acetone cooled condensor (V-shaped cold finger-type) and a secondary in-line vacuum trap (dry ice-acetone cooling) to collect solvent. The checkers used a diaphragm-type vacuum pump equipped with −20 °C cooling circulator.
7.
Further purification was not necessary; material obtained by bulb-to-bulb distillation was 99.5% pure (GC), and was used in the subsequent step. GC retention time of 11.90 min (capillary GC, HP-5 column, 0.25 µm × 30 m; temperature program: 40 °C for 4 min, then increased 25 °C/min to 280 °C. To obtain an accurate boiling point, the ester was distilled at 80–81 °C (1.1 mmHg) through a short path column. Analytical data is reported for the distillate obtained from the bulb-to-bulb distillation: Analytical TLC: R
f 0.36 (10 %
ethyl acetate in hexanes);
1H NMR
pdf (500 MHz, CDCl
3) δ: 2.53 (t,
J = 2.4 Hz, 1 H), 4.92 (d,
J = 2.4 Hz, 2 H), 7.46 (t,
J = 7.9 Hz, 2 H), 7.55 (tt,
J = 7.4, 1.2 Hz, 1 H), 8.06–8.08 (m, 2 H);
13C NMR
pdf (125 MHz, CDCl
3) δ: 52.4, 75.0, 77.7, 128.4, 129.3, 129.7, 133.3, 165.7; FT-IR (thin film, cm
−1) 3296, 1725, 1268, 1108; HRMS (FAB; M
+ + H,
m/z) calcd for C
10H
9O
2 161.0602, found 161.0604.
8.
The Schlenk tube dimensions were 3.7 cm × 18 cm with a sidearm of 9 mm OD. The septum was fitted over the 9 mm tube with argon supplied through an 18-gauge needle. The teflon cannula is threaded through the same septum. Use of a round-bottomed flask in place of the Schlenk tube resulted in incomplete conversion.
9.
The submitters reported the use of a cold-finger condenser in the Schlenk tube. The checkers used a standard reflux condenser and obtained identical results.
10.
All-
cis polybutadiene (Mw ca. 2–3 × 106, 98% cis) was purchased from Aldrich (18137–4) and used without further purification. The polymer was kept in a polyethylene bag and no special precautions were taken to exclude air. The use of fewer equivalents of
polybutadiene led to incomplete conversions. For example, 4.0 equiv of polybutadiene delivered under the same reaction conditions described in this procedure produced only
64% yield of
cyclohexadiene (NMR yield against
mesitylene internal standard).
11.
Ruthenium [1,3-bis-(2,4,6-trimethylphenyl)-2-imidazolidinylidine]-dichloro(phenylmethylene)(tricyclohexylphosphine) (Grubbs second generation catalyst) was obtained from Aldrich Chemical Co. (Milwaukee, WI) or from Materia, Inc. (Pasadena, CA).
12.
A Teflon cannula was used (ID = 0.8 mm and 0.4 mm thickness of about 20 cm length). One end of the cannula was attached to the needle by slipping over the 22 gauge stainless steel needle until snug; the other end was cut with a razor blade at a 45° angle and pierced through the rubber septum. The tip of the cannula was touched to the cooled glass surface just below the level of solvent reflux.
13.
Ethyl vinyl ether (99%) was used as received from Aldrich Chemical Co.14.
The filtration was carried out using Merck silica gel 60 (230–400 mesh). The viscous crude mixture required high pressure to elute the solvent. The use of a smaller column (<2 cm) was not suitable.
15.
This fraction contains oligomeric byproducts including isomeric 1,5,9-cyclododecatrienes. This fraction should be checked for
cyclohexadiene product by TLC analysis (R
f 0.42;
10% ethyl acetate in hexanes) before discarding.
16.
The submitters reported obtaining a pale yellow oil. The
cyclohexadiene is not stable as the neat liquid, even when stored at −20 °C. Partial decomposition by polymerization was variable, and depended significantly on sample storage conditions. When BHT (5 % by weight) was added to the distillate, polymerization was retarded somewhat. Extended storage of the neat
cyclohexadiene is not recommended. Pure cyclohexadiene could be obtained from degraded samples by redistillation with loss of material.
17.
Further purification was not necessary. In order to obtain an accurate boiling point, the dienylester was distilled at 124–125 °C (1.1 mmHg) through a short path column.
18.
The analytical data are provided for the distillate from the bulb-to-bulb distillation: Analytical TLC: R
f 0.42 (10 %
ethyl acetate in hexanes).
1H-NMR
pdf (500 MHz, CDCl
3) δ: 2.15–2.26 (m, 4 H), 4.80 (m, 2 H), 5.87–5.93 (m, 2 H), 5.99 (ddd,
J = 9.5, 3.4, 1.5 Hz, 1 H), 7.44 (t,
J = 7.6 Hz, 2 H), 7.55 (tt,
J = 7.4, 1.2 Hz, 1 H), 8.05–8.08 (m, 2 H);
13C-NMR
pdf (125 MHz, CDCl) δ: 22.0, 22.1, 67.0, 124.3, 124.9, 127.6, 128.3, 129.6, 130.3, 131.1, 132.9, 166.5; FT-IR (thin film, cm
−1) 2935, 1719, 1450, 1271, 1111, 712; HRMS (FAB; M
+,
m/z) calcd for C
14H
15O
2 215.1072, found 215.1069. GC was used to assess purity of the
cyclohexadiene, which was found to be >98.5%. GC retention time of 15.07 min (capilliary GC, HP-5 column, 0.25 µm × 30 m; temperature program 40 °C for 5 minutes, then increased 25 °C/min to 280 °C).
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
In recent years, alkene metathesis has emerged as a powerful method for the synthesis of the carbon-carbon double bond. Synthetic applications increased dramatically with the advent of the functional group tolerant ruthenium carbenes developed by Grubbs.
3,4,5,6,7,8,9,10,11 Enyne metathesis is a closely related catalytic reaction which involves the coupling of an alkene with an alkyne to give a conjugated 1,3-diene.
12 This is a particularly attractive reaction because the conjugated diene product participates in a host of transformations and can thus be used as a building block for the synthesis of more complex organic molecules.
Recent metathesis applications involving tandem transformations are exciting due to their potential to build rings from simple acyclic precursors. These applications are very attractive because they permit the rapid assembly of complex structures. Tandem metathesis transformations had previously been limited to intramolecular reactions.
13,14 Though attractive, these tandem reactions require the synthesis of polyenyne reactants. Ring synthesis from unsaturated reactants by an
intermolecular reaction is advantageous because the reactants are comparatively simple. The difficulty in intermolecular ring building reactions is the initial, non-stereoselective metathesis between the alkene and alkyne component. Control of alkene geometry is a current, general problem in intermolecular metathesis research. The tandem diene-alkyne metathesis described in this procedure is challenging because it is triggered by an intermolecular bond formation which lacks stereocontrol.
The challenge posed by nonstereoselective cross metathesis was addressed by development of "methylene-free" conditions. In general, the low stereoselectivity in cross enyne metathesis of alkenes and alkynes limits its utility in synthesis.
15 Low selectivity was also evident in our initial work on the tandem dienyne metathesis.
16 In the case of
1,3-cyclohexadiene synthesis, only the
Z-alkene would lead to the ring-closed product. This problem led us to consider interconversion of vinyl carbene intermediates to produce more of the
Z-intermediate, which would then give ring-closing metathesis. This process would produce more of the desired cyclohexadiene product. Our analysis led us to consider conditions that would favor interconversion of vinyl carbenes by omitting CH
2 sources. These conditions were expected to decelerate the rate of catalyst turnover, giving the isomeric vinyl carbene intermediates time to interconvert. Since there are no CH
2 sources in the reaction and no L
nRu = CH
2 intermediates, we termed these conditions to be “methylene-free.”
2 The term is used to differentiate these conditions from typical cross metatheses, which had previously required the use of 1-alkene reactants. (However, the catalytic enyne metathesis mechanism is complex,
17,18,19 and the hypothesis that methylene-free conditions permit the equilibration of vinyl carbene intermediates requires further experimentation.)
The procedure described here illustrates that the methylene-free conditions provide an effective ring synthesis of 1,3-cyclohexadienes. In the best cases, the cyclohexadienes are produced in greater than 80% yield (as assessed by NMR). The isolation requires quenching of carbene reactivity and separation of oligomeric materials derived from polybutadiene. In this procedure, carbene reactivity is quenched upon addition of ethyl vinyl ether (EVE). However, our group has recently developed an improved protocol for the quenching and removal of ruthenium carbene, which can also be used in the methylene-free synthesis of 1,3-cyclohexadienes (see eq 8, Scheme 3 in ref. 20).
20 In terms of alkene reactant, the procedure employs polybutadiene (PB) as the alkene reactant, yet similar results are obtained using
1,5-cyclooctadiene (1,5-COD) as the alkene. In the 20-mmole scale procedure above using 4 equiv of 1,5-COD (8 alkene equivalents),
61% yield of the corresponding
1,3-cyclohexadiene was obtained (in comparison to
65% with 8 equiv PB). However, use of fewer equivalents of
polybutadiene (4 equiv) led to incomplete conversion of alkyne reactant (
64% conversion of
propargyl benzoate as assessed by
1H NMR comparison to the mesitylene internal standard).
Table 1 shows results using 4 equiv of 1,5-COD in place of
polybutadiene on a 1-mmole scale. On the smaller scale, catalyst loading was not optimized lower than 5 mole %, though it is anticipated that catalyst loading can be decreased during scale-up.
21 We have found that either 8 equiv PB or 4 equiv 1,5-COD can be used with equal efficiency.
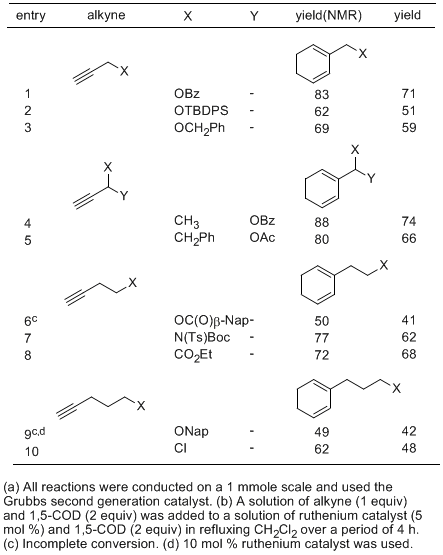
A variety of terminal alkynes participated efficiently in this reaction. Esters at the propargylic and homopropargylic position were well tolerated (entries 1, 4, 5, 6). Protected propargyl ethers gave reasonable isolated yields (entries 2, 3). Nitrogen-containing homopropargylic functionality was tolerated if suitably protected (entry 7), and a remote ester posed no difficulty (entry 8). The more remote naphthoate ester required higher catalyst loading and still proceeded to only partial conversion (entry 9). This is presumably due to a coordination problem in the intermediate vinyl carbenes, which may both slow the reaction and result in catalyst decomposition. Coordinating ethers in the homopropargylic position were similarly not well tolerated under the conditions reported in this procedure; however, conditions using higher concentrations of 1,5-COD have been successful with these substrates.
2b Since internal alkenes coordinate more weakly to metals than 1-alkenes, the putative slow step, vinyl carbene reaction with alkene, is slower than a typical enyne metathesis involving 1-alkenes. As a result, the reaction proves more sensitive to potentially coordinating functional groups than typical ring-closing alkene or enyne metathesis reactions. In these unusual cases, higher catalyst loading and higher alkene concentrations are often necessary. With higher concentration of alkene (polybutadiene or 1,5-COD), internal alkynes have been successfully used in the 1,3-cyclohexadiene ring synthesis.
2b Methylene-free conditions have also been used for the ring expansion of cyclopentene to give 1,3-cycloheptadienes.
22In conclusion, a practical synthesis of 1,3-cyclohexadienes is possible from simple, readily available unsaturated reactants. The optimal 20-mmole scale procedure employs the second generation Grubbs carbene as a functional group tolerant catalyst. A moderately low catalyst loading was achieved despite the demands of a slow intermolecular reaction. The key to this method is the use of methylene-free conditions, achieved with either polybutadiene or 1,5-cyclooctadiene as the alkene.
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
Propargyl alcohol:
2-Propyn-1-ol; (107-19-7)
Benzoyl chloride; (98-88-4)
Triethylamine:
N,N-Diethylethanamine; (121-44-8)
DMAP: 4-Pyridinamine, N,N-dimethyl-; (1122-58-3)
Propargyl benzoate:
2-Propyn-1-ol, benzoate; (6750-04-5)
cis-Polybutadiene; (40022-03-5)
Grubbs' second generation catalyst: Ruthenium [1,3-bis-(2,4,6-trimethylphenyl)-2-imidazolidinylidine]-dichloro(phenylmethylene)(tricyclohexylphosphine); (246047-72-3)
Ethyl vinyl ether:
Ethene, ethoxy-; (109-92-2)
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved