Org. Synth. 2007, 84, 43
DOI: 10.15227/orgsyn.084.0043
STEREOSELECTIVE ISOPRENOID CHAIN EXTENSION WITH ACETOACETATE DIANION:
[(E, E, E)-GERANYLGERANIOL FROM (E, E)-FARNESOL]
Submitted by Yinghua Jin, Frank G. Roberts, and Robert M. Coates
1.
Checked by Hillary M. Peltier and Jonathan A. Ellman.
1. Procedure
A. (E, E)-Farnesyl bromide. A flame-dried, 500-mL, three-necked, round-bottomed flask, equipped with a magnetic stirring bar, two rubber septa, and a nitrogen inlet connected to a mineral oil bubbler, is charged with 10.0 g (11.3 mL, 45.0 mmol) of (E, E)-farnesol (Note 1) and 150 mL of THF (Note 2). The solution is stirred and cooled in a dry ice/acetonitrile bath (temperature −45 °C to −47 °C), while methanesulfonyl chloride (6.70 g, 4.53 mL, 58.5 mmol) (Note 3) is added through one septum via syringe over 5 min. Triethylamine (9.10 g, 12.6 mL, 89.9 mmol) (Note 4) is then added through one septum by syringe over 5 min. A white solid precipitates during the addition, and the resulting suspension is stirred at −45 °C to −47 °C for 45 min. A room temperature solution of lithium bromide (15.6 g, 180 mmol) (Note 5) in 50 mL of THF is prepared in a separate flask and added by cannula transfer over 5 min. The dry ice/acetonitrile bath is removed, and the suspension is allowed to warm to 0 °C with an ice-bath. The yellowish mixture is stirred at 0°C for 1 h. The reaction mixture is poured into 300 mL of ice water. The aqueous layer is separated and extracted with three 150-mL portions of cold diethyl ether (Note 6). The combined organic extracts are sequentially washed with ice-cold saturated NaHCO3 (200 mL) and brine (200 mL), dried over anhydrous MgSO4 (Note 7), filtered, and concentrated by rotary evaporation and vacuum drying to give 11.6 g of (E, E)-farnesyl bromide as a light yellow oil which is used in the next step without purification (Note 8).
B. (6E, 10E)-Ethyl 7,11,15-Trimethyl-3-oxohexa-deca-6,10,14-trienoate. A flame-dried, 500-mL, three-necked, round-bottomed flask, equipped with a magnetic stirring bar, two rubber septa, and a nitrogen inlet connected to a mineral oil bubbler, is charged with a 60% dispersion of sodium hydride in mineral oil (5.32 g, 133 mmol) (Note 9). The mineral oil is removed by washing with three 10-mL portions of hexanes, allowing the sodium hydride to settle and withdrawing the supernatant solvent with a syringe. Tetrahydrofuran (100 mL) (Note 2) is added, and the hydride suspension is stirred and cooled in a 0 °C ice-bath as ethyl acetoacetate (15.8 g, 15.5 mL, 121 mmol) (Note 10) is added dropwise via syringe over 15 min. The light yellow solution of the sodium enolate is stirred at 0 °C for an additional 15 min (Note 11). A solution of n-BuLi in hexane (59.2 mL, 2.15 M, 127 mmol) (Note 12) is then added by syringe over 15 min. The resulting orange solution of acetoacetate dianion is stirred at 0 °C for 15 min. A room temperature solution of farnesyl bromide (11.6 g, 40.6 mmol) in 20 mL of THF is added by cannula transfer over 10 min. The resulting orange suspension is stirred at 0 °C for 15 min, aqueous HCl (1N, 300 mL) is added, and the aqueous layer is separated and extracted with diethyl ether (3 × 150 mL). The combined organic extracts are washed sequentially with saturated aqueous NaHCO3 (2 × 200 mL) and brine (2 × 200 mL), dried over anhydrous MgSO4, filtered, and concentrated by rotary evaporation and vacuum drying to give 21.6 g of crude product. The excess ethyl acetoacetate is removed by Kugelrohr distillation (2 h, 60 °C, 0.4 mmHg) to afford 16.0 g of β-keto ester 3 as an orange oil. The residue is loaded onto a column of 220 g of silica gel (Note 13) and eluted with 300 mL of hexanes. At that point, collection of 50-mL fractions in glass tubes (25 × 150 mm) is begun, while elution is continued with 500 mL of 2% EtOAc-hexanes, 500 mL of 5% EtOAc-hexanes, 500 mL of 10% EtOAc-hexanes and then 500 mL of 15 % EtOAc-hexanes. The fractions are analyzed by TLC on silica gel with 30% EtOAc-hexanes as eluent and visualized with p-anisaldehyde staining reagent (Note 14). The product is obtained in fractions 20–33, which are combined and concentrated under reduced pressure using a rotary evaporator to yield β-keto ester 3 (11.8 g, overall 78% from farnesol) as a light yellow oil (Note 15).
C. (2Z,6E,10E)-Ethyl 7,11,15-Trimethyl-3-diethylphosphoryloxy-6,10,14-trienoate. A flame-dried, 500-mL, three-necked, round-bottomed flask, equipped with a magnetic stirring bar, two rubber septa, and a nitrogen inlet connected to a mineral oil bubbler, is charged with a 60% dispersion of sodium hydride in mineral oil (1.62 g, 40.6 mmol) (Note 9). The mineral oil is removed by washing with hexanes (3 × 10 mL) allowing the sodium hydride to settle and withdrawing the supernatant solvent with a syringe. Diethyl ether (150 mL) (Note 16) is added, and the mixture is stirred and cooled at 0 °C with an ice-bath. β-Keto ester 3 (11.7 g, 35.3 mmol) in 50 mL of diethyl ether is added slowly via cannula over 20 min, and the reaction mixture is stirred at rt for 20 min or until gas evolution ceases. The clear orange solution is then cooled to 0 °C, and neat diethyl chlorophosphate (9.13 g, 7.67 mL, 52.9 mmol) (Note 17) is added by syringe over 5 min. The mixture is stirred at 0 °C for 15 min, after which time the reaction is quenched by adding saturated aqueous NH4Cl (300 mL). The aqueous layer is separated and extracted with diethyl ether (3 × 150 mL). The combined organic extracts are washed sequentially with saturated aqueous NaHCO3 (2 × 200 mL) and brine (2 × 200 mL), dried over anhydrous MgSO4, filtered, and concentrated by rotary evaporator and vacuum drying to give the crude enol phosphate 4 (16.9 g) as a yellow oil (Note 18).
D. (2E,6E,10E)-Ethyl 3,7,11,15-Tetramethylhexadeca-2,6,10,14-tetraenoate. A flame-dried, 500-mL three-necked, round-bottomed flask, equipped with a magnetic stirring bar, one rubber septum, a thermometer, and a nitrogen inlet connected to a mineral oil bubbler, is charged with copper(I) iodide (12.1 g, 63.5 mmol) (Note 19) and 40 mL of diethyl ether (Note 16). The suspension is stirred and cooled at 0 °C in an ice bath as methyllithium in diethyl ether (98.8 mL, 1.29 M, 127 mmol) (Note 20) is added via syringe over 5 min. A yellow solid precipitates immediately during the addition, and the precipitate disappears after stirring at 0 °C for 30 min. The colorless or light tan solution is cooled to −78 °C using a dry ice/acetone bath and stirred magnetically, as a room temperature solution of enol phosphate 4 (16.9 g) in 50 mL of diethyl ether is added slowly over 20 min via cannula while maintaining the internal temperature below −60 °C (Note 21). The orange-red solution is stirred at −78 °C for 2 h. The reaction mixture is then allowed to warm to −45 °C using a dry ice/acetonitrile bath and stirred for 2 h, resulting in a dark purple mixture. Methyl iodide (4.50 mL, 10.3 g, 72.3 mmol) is added while stirring at −45 °C for 10 min (Note 22). The mixture is then poured into 300 mL of ice-cold 4:1 (v:v) saturated NH4Cl and concentrated NH4OH mixture, and stirred for approximately 1 h until all the copper salts disappear. The aqueous layer is separated and extracted with ether (3 × 150 mL). The combined organic extracts are washed sequentially with 10 % NH4OH (2 × 200 mL), water (2 × 200 mL), and brine (2 × 200 mL), dried over anhydrous MgSO4, filtered, and concentrated by rotary evaporation and vacuum drying to give crude ester 5 (9.58 g) as a 19:1–20:1 mixture of trans and cis isomers according to GC analysis (Note 23).
E. (E, E, E)-Geranylgeraniol. A flame-dried, 500-mL, three-necked, round-bottomed flask, equipped with a magnetic stirring bar, two rubber septa, and a nitrogen inlet connected to a mineral oil bubbler, is charged with ester 5 (9.58 g, ~ 28.8 mmol) and toluene (100 mL) (Note 24). The solution is cooled in a dry ice/acetone bath (temperature −78 °C), and diisobutylaluminum hydride in hexanes (86.4 mL, 86.4 mmol) (Note 25) is added slowly via syringe through one of the septa over 20 min. After 30 min at −78 °C, the reaction is quenched by careful addition of methanol (10 mL) via a glass pipette over approximately 10 min (Note 26). The mixture is allowed to warm to room temperature and poured into a mixture of saturated NH4Cl (250 mL) and 1N HCl (250 mL) with stirring, and the stirring is continued for 30 min. The aqueous layer is separated and extracted with ether (3 × 200 mL). The combined organic extracts are sequentially washed with water (2 × 200 mL) and brine (2 × 200 mL), dried over anhydrous MgSO4, filtered, and concentrated by rotary evaporator and vacuum drying. The concentrated residue is loaded onto a column (5.0 × 21 cm) of 200 g of silica gel (Note 13) and eluted with 300 mL of hexanes. At that point, collection of 50-mL fractions in glass tubes (25 × 150 mm) is begun, while elution is continued with 500 mL of 5% EtOAc-hexanes, 500 mL of 10% EtOAc-hexanes, 500 mL of 15% EtOAc-hexanes, and then 500 mL of 20 % EtOAc-hexanes. The fractions are analyzed by TLC on silica gel with 30% EtOAc-hexanes as eluent and visualized with p-anisaldehyde staining reagent (Note 14). The product is obtained in fractions 24–42, which are combined and concentrated under reduced pressure using a rotary evaporator to yield geranylgeraniol (7.20 g, overall 70% from β-keto ester 3) (Note 27).
2. Notes
1.
Trans,
trans-farnesol (~95%) was purchased from the Fluka Chemical Corp. and was used without further purification.
2.
Tetrahydrofuran was purchased from Fisher Scientific Co. and passed through an alumina column (checkers) or purchased from Burdick and Jackson and freshly distilled from sodium/benzophenone under nitrogen (submitters).
3.
Methanesulfonyl chloride (>99.5 % purity) was purchased from Aldrich Chemical Co. and used as received. When this reagent was taken from a previously opened container, it was distilled from P
2O
5 before use.
4.
Triethylamine (reagent grade) was purchased from Fisher Scientific Co., distilled from calcium hydride, and stored over potassium hydroxide pellets.
5.
Lithium bromide was purchased from Fisher Scientific Co. and flame-dried under vacuum before use.
6.
Unless otherwise indicated, the solvents used were of reagent grade quality and were used without further purification.
7.
Anhydrous MgSO
4 was purchased from Fisher Scientific Co.
8.
Bromide
2 is prone to decomposition and should be kept cold by adding ice during the extractions and washings. It should be stored in a freezer (approximately −20 °C) no longer than one day. The physical data for (
E,
E)-farnesyl bromide (provided by submitters) follow: TLC R
f = 0.72 (7:3 hexanes/ethyl acetate);
1H NMR
pdf (500 MHz, CDCl
3) δ: 1.61 (br s, 6 H, 2C
H3), 1.69 (s, 3 H, C
H3), 1.74 (s, 3 H, C
H3), 1.97–2.13 (m, 8 H, 4C
H2), 4.03 (d,
J = 8.3 Hz, 2 H, C
H2Br), 5.08–5.11 (m, 2 H, vinyl
H), 5.54 (br t,
J = 8.3 Hz, 1 H, vinyl
H);
13C NMR (100 MHz, CDCl
3) δ: 16.2, 16.3, 17.9, 25.9, 26.3, 26.9, 30.0, 39.7, 39.9, 120.7, 123.6, 124.5, 131.6, 135.8, 143.8.
9.
Sodium hydride was purchased from Acros Organics and used as received.
10.
Ethyl acetoacetate was purchased from Aldrich Chemical Co. and purified by washing with saturated aqueous NaHCO
3 and water, drying over MgSO
4, and distilling under reduced pressure (bp 30–32 °C, 1 mmHg). A slower, incomplete reaction (89 % conversion after 5 h at 0 °C, 68% yield based on conversion) occurred when only 1.2 equiv of ethyl acetoacetate was used, instead of 3.0 equiv, under otherwise identical conditions.
11.
Caution: Vigorous evolution of hydrogen occurs. The mixture should be stirred until gas evolution ceases.12.
n-Butyllithium in hexanes was purchased from Aldrich Chemical Co. and titrated using 2,6-di-
tert-butyl-4-methylphenol and 1,10-phenanthroline (checkers).
13.
Silica gel 60 230–400 mesh ASTM was purchased from Merck and Co. The 6.2-cm diameter glass column was packed with silica gel (6.2 × 14.5 cm) and hexanes.
14.
Anisaldehyde staining reagent was prepared by adding, in small portions with stirring, 6.5 mL of concentrated H
2SO
4, 2.5 mL of glacial acetic acid, and 5 mL of
p-anisaldehyde in the order listed to 186 mL of cold 95% EtOH.
15.
The absolute yield of β-keto ester
3 was determined in one of the submitters' runs prior to chromatography (17.3 g of crude
3) by
1H NMR analysis using mesitylene as a standard. A solution of crude product (47 mg) and mesitylene (11 mg) in chloroform-
d was prepared. Integrations of the
1H NMR spectrum showed a 1.00:0.79 molar ratio of the product and the standard, indicating the yield was 13.7 g (91 %). The submitters obtained 11.7–12.2 g of β-keto ester
3 (overall 78–81% from farnesol) after chromatography as mixtures of keto and enol tautomeric forms. The physical data (supplied by the checkers) for pure β-keto ester
3 (6.7:1 mixture of keto and enol tautomeric forms) are as follows: TLC: R
f = 0.60 (7:3, hexanes/ethyl acetate); IR: 1721, 1745, 2920 cm
−1.
1H NMR
pdf (500 MHz, CDCl
3) δ: 1.27 (t,
J = 7.2 Hz, 3 H, C
H3), 1.59 (br s, 3 H, C
H3), 1.60 (s, 3 H, C
H3), 1.61 (s, 3 H, C
H3), 1.68 (br s, 3 H, C
H3), 2.00–2.13 (m, 8 H, 4 C
H2), 2.28 (q,
J = 7.5 Hz, 2 H, C
H2), 2.56 (t,
J = 7.5, 2 H, C
H2), 3.42 (s, 2 H, C
H2), 4.19 (q,
J = 7.2 Hz, 2 H, C
H2), 4.97 (s, 0.164 H, -C(OH)=C
H-), 5.06–5.09 (m, 3 H, vinyl
H), 12.1 (s, 0.146 H, -C(O
H)=CH-).;
13C NMR (125 MHz, CDCl
3) δ: 14.4, 16.33, 16.35, 18.0, 22.5, 26.0, 26.9, 27.1, 39.9, 40.0, 43.4, 49.7, 61.7, 122.4, 124.4, 124.7, 131.6, 135.4, 137.1, 167.6, 202.9. Anal. Calcd. for C
21H
34O
3: C, 75.41; H, 10.25. Found: C, 75.63; H, 10.47.
16.
Diethyl ether was purchased from Fisher Scientific Co. and passed through an alumina column (checkers) or freshly distilled from sodium/benzophenone under nitrogen (submitters).
17.
Diethyl chlorophosphate (97%) was purchased from Aldrich Chemical Co. and was used as received.
18.
The submitters recommend using the unpurified enol phosphate since an attempt to purify the intermediate by large-scale flash column chromatography resulted in reduced yield, probably owing to the decomposition of the product. The crude enol phosphate
4 (18.7 g) was analyzed by the submitters by
1H NMR spectroscopy using mesitylene as a standard. A solution of 20 mg of enol phosphate
4 and 10 mg of mesitylene in chloroform-
d was prepared. Integrations of the
1H NMR spectrum showed a 1.00:2.08 molar ratio of the product and the standard, indicating the purity of the crude product was 90 %. A 70-mg sample of enol phosphate was purified by the submitters with flash column chromatography using 30 % ethyl acetate in hexanes as eluent to yield 27 mg of pure enol phosphate
4: TLC: R
f = 0.26 (7:3 hexanes/ethyl acetate);
1H NMR
pdf (400 MHz, CDCl
3) δ: 1.27 (t,
J = 7.1 Hz, 3 H, C
H3), 1.36 (td,
J = 7.1, 1.2 Hz, 6 H, 2C
H3), 1.59 (s, 3 H, C
H3), 1.60 (s, 3 H, C
H3), 1.61 (s, 3 H, C
H3), 1.68 (s, 3 H, C
H3), 1.95–2.07 (m, 8 H, 4C
H2), 2.27 (q,
J = 7.3 Hz, 2 H, C
H2), 2.45 (t,
J = 7.6 Hz, 2 H, C
H2), 4.15 (q,
J = 7.2 Hz, 2 H, C
H2), 4.26 (dq,
J = 7.8, 7.1 Hz, 4 H, 2C
H2), 5.07–5.11 (m, 3 H, vinyl
H), 5.35 (s, 1 H, vinyl
H);
13C NMR (CDCl
3, 125 MHz) δ: 14.4, 16.16, 16.23, 16.28, 16.29, 17.9, 25.1, 25.9, 26.8, 26.9, 35.4, 39.9 (d,
J = 7.6 Hz), 60.1, 64.9 (d,
J = 6.1 Hz), 105.5 (d,
J = 7.4 Hz), 122.0, 124.2, 124.5, 131.5, 135.3, 137.0, 161.6 (d,
J = 7.3 Hz), 164.0;
31P NMR (CDCl
3, 202 MHz) δ −7.6.
19.
Copper(I) iodide (98%) purchased from Aldrich Chemical Co. was used as received.
20.
Methyllithium in diethyl ether was purchased from Aldrich Chemical Co. and titrated using 2,6-di-
tert-butyl-4-methylphenol and 1,10-phenanthroline (checkers).
21.
Enol phosphate
4 should be added slowly. The coupling reaction was exothermic; fast addition of enol phosphate
4 caused a temperature rise that led to a lower
trans/
cis ratio.
22.
Direct hydrolysis of the crude product with a mixture of ice-cold 5:1 (
v/
v) saturated NH
4Cl and concentrated NH
4OH afforded all
trans ester
5 contaminated by approximately 5% of the simple reduction byproduct, (2
E,6
E,10
E) ethyl 7,11,15-trimethylhexadeca-2,6,10,14-tetraenoate, which was inseparable by chromatography. Prior addition of methyl iodide decreased the amount of the reduction byproduct to ≤ 1%.
23.
The unpurified ester
5 (11.1 g) was analyzed by the submitters in one run by
1H NMR spectroscopy using mesitylene as internal standard. Integrations of the spectrum of the ester (26 mg) and mesitylene (10 mg) in chloroform-
d showed a 1.00:1.17 molar ratio of the product and standard, indicating a purity of 90 %. In a different run a small sample (approximately 30 mg) of the crude product was purified by the submitters with flash column chromatography using 10 % ethyl acetate in hexanes as eluent to afford a characterization sample of pure ester
5: TLC R
f = 0.72 (7:3 hexane/ethyl acetate);
1H NMR
pdf (400 MHz, CDCl
3) δ: 1.28 (t,
J = 7.2 Hz, 3 H, C
H3), 1.60–1.61 (m, 9 H, 3C
H3), 1.69 (d,
J = 1.1 Hz, 3 H, C
H3), 1.96–2.17 (m, 12 H, 6C
H2), 2.17 (d,
J = 1.1 Hz, 3 H, C
H3), 4.15 (q,
J = 7.2 Hz, 2 H, C
H2), 5.08–5.13 (m, 3 H, vinyl
H), 5.67 (m, 1 H, vinyl
H);
13C NMR (100 MHz, CDCl
3) δ: 14.6, 16.22, 16.25, 17.9, 19.0, 25.9, 26.2, 26.8, 27.0, 39.88, 39.92, 41.2, 59.7, 115.8, 123.1, 124.3, 124.6, 131.5, 135.3, 136.4, 160.1, 167.1.
24.
Toluene was purchased from Fisher Scientific Co. and passed through a column of alumina (checkers) or dried over 4Å molecular sieves (submitters).
25.
Diisobutylaluminum hydride (1.0 M in hexanes) was purchased from Aldrich Chemical Co.
26.
Caution: Methanol should be added very slowly to avoid vigorous gas evolution.27.
The submitters obtained 6.90–7.94 g of geranylgeraniol (68–77% yield from β-keto ester
3). Physical data for geranylgeraniol (
6) (supplied by the checkers): TLC: R
f = 0.43 (7:3 hexanes/ethyl acetate); IR: 1445, 1668, 2921, 3319 cm
−1.
1H NMR
pdf (500 MHz, CDCl
3) δ: 1.59 (br s, 9 H, 3C
H3), 1.67 (br s, 6 H, 2C
H3), 1.98–2.13 (m, 12 H, 6C
H2), 4.13 (d, 2 H,
J = 7.0), 5.07–5.10 (m, 3 H, vinyl
H), 5.40 (t,
J = 7.0 Hz, 1 H, vinyl
H);
13C NMR (CDCl
3, 125 MHz) δ: 16.2, 16.3, 16.6, 18.0, 26.0, 26.6, 26.9, 27.0, 39.9, 39.99, 40.0, 59.6, 123.7, 124.1, 124.5, 124.7, 131.5, 135.2, 135.6, 139.9. Anal. Calcd. for C
20H
34O: C, 82.69; H, 11.80. Found: C, 82.58; H, 12.16. The purity of geranylgeraniol (
6) was determined to be 96 % according to GC analysis. GC analysis was performed on a Shimadzu GC-14A with 30-m Restek Rtx-5 column, and the sample was analyzed using the following parameters, initial temperature 40 °C, hold for 5 min, 20 °C/min until 300 °C, hold for 5 min.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
(
E, E,
E)-Geranylgeranyl diphosphate (GGPP) is considered to be the universal substrate for diterpene cyclases, prephytoene synthase, and many prenyl diphosphate synthases,
2 as well as cosubstrate for protein geranylgeranyl transferases I and II.
3 The diphosphate ester is readily prepared from (
E, E,
E)-geranylgeraniol (
6, GGOH) by conversion to the corresponding allylic chloride, bromide, or diethyl phosphate followed by
SN2 displacement with tetrabutylammonium pyrophosphate.
4,5 Although both GGOH and GGPP are commercially available, the prices are quite high, and there is need for reliable methods to access different labeled forms and analogues as pure all trans isomers for research on diterpene synthases and transferases. The present five-step procedure (
1 →
6) effects isoprene chain extension of (
E,
E)-farnesol to give 6.9–7.9 g of the isoprenylogue
6 in 53–61% overall yield and with high E selectivity using Weiler's acetoacetate dianion alkylation-enol phosphate coupling methods.
6,7 6-FluoroGGOH was prepared in the same way from 2-fluorofarnesol.
8 A similar route to
6 utilizes a Suzuki reaction of the enol triflate related to enol phosphate
4 to obtain α,β-enoate
5.
9The most common syntheses of (
E, E,
E)-geranylgeraniol (
6) in the literature proceed by C
2 chain extensions of (
E,
E)-farnesylacetone, reactions that invariably afford mixtures of 2E and 2Z isomers of esters or alcohols. Farnesylacetone has been synthesized from farnesol derivatives by traditional α-alkylation of acetoacetate
10a,b and by alkylation of lithiated acetone
N,
N-dimethylhydrazone,
10c and by Wittig olefination of the keto aldehyde generated from ozonolysis of isoprene cyclic trimer.
11 Conversion to isomeric mixtures of geranylgeranoates or geranylgeraniol has been accomplished by an acetylide addition-reduction-allylic rearrangement sequence,
10a by Horner-Wadsworth-Emmons olefinations,
12 by ethoxyacetylene addition and hydrolysis,
10b,13 and by vinylmetal addition followed by allylic rearrangement.
10c,11 Reductions of ester
5 to the GGOH have been carried out with LiAlH
4,
iBu
2AlH, and LiAlH
4-AlCl
3.
Geranylgeraniol has also been obtained by Peterson and Wittig olefinations to complete the isoprene units at the opposite ends of the chain.
14 A convergent approach involves allylic couplings of two C
10 geraniol derivatives,
15 geranylbarium alkylation,
16a and an allyl silane-acetal
SE' method.
16b Palladium-mediated couplings of an α-sulfonylfarnesyl-1-carboxylate with isoprene 3,4-epoxide and of methyl farnesoate with α-sulfonyl-γ,γ-dimethylallylcarboxylate have been reported.
17 Synthesis of the 2E, 6Z, 10E isomer has been achieved by sequential Pd-catalyzed couplings of zincates with vinyl iodides followed by zirconocene-mediated addition of Me
3Al to a trienyne intermediate to form the hydroxyl-bearing isoprene unit.
18 An iterative C5 isoprenoid chain extension approach to (
E, E, E,)geranylgeraniol by means of Biellmann couplings with sulfonyl-stabilized anions has been reported recently.
19a A moderately selective method (E/Z 91:9) for effecting palladium-catalyzed allylic rearrangement of geranyllinalyl acetate followed by cleavage of the resulting amine to geranylgernaiol has been disclosed.
19b Patents describe the separation of geranylgeranyl sterate isomers by low temperature crystallizations,
19c and isolation of high purity (
E, E, E)-geranylgeraniol after hydrolysis of naturally occurring esters extracted from annatto seeds.
19dAlthough numerous methods for isoprenoid chain extensions have been investigated,
20 the present approach
6a,b,7 has the important advantages of high E-selectivity at both the 2,3 and 6,7 double bonds, satisfactory overall yield, and the potential to form 2Z,6E, 2E,6Z, and 2Z,6Z isomers.
21 The 5-step sequence begins with conversion of (
E,
E)-farnesol (
1) to the corresponding bromide (
2) with methanesulfonyl chloride, triethylamine, and lithium bromide in THF at −45 to 0 °C.
22 The Li/Na dianion is generated by lithiation of sodio acetoacetate with
n-butyllithium in THF at 0 °C, and subsequent γ-alkylation with the unpurified bromide affords β-keto ester
3. Phosphorylation of the sodium enolate with diethyl chlorophosphate in ether gives rise to all trans enol phosphate
4 which undergoes vinylic coupling with lithiodimethyl cuprate in ether solution at −78 to −45 °C to give (
E,
E,
E)-ethyl geranylgeranoate (
5) in good yield and ~20:1 E-selectivity. Reduction of the ester with isobutylaluminum hydride in toluene provides (
E,E,
E)-geranylgeraniol (
6) in 53–61% overall yield after purification by chromatography on silica gel.
Acetoacetate dianion is a very useful carbon nucleophile for C-C bond formation at the γ-position and for accessing a wide variety of β-keto esters.
23 It is sufficiently stable to undergo alkylations with unactivated primary halides and epoxides,
24 and aziridines bearing
N-sulfonyl or
N-phosphoryl groups.
25 Reactions with carbonyl electrophiles afford δ-hydroxy-β-keto esters and α, β-diketo esters (Scheme 1).
26Scheme 1
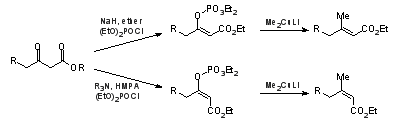
Coupling of enol phosphates with organocopper reagents offers a versatile, stereoselective synthesis of tri- and tetra-substituted α,β-unsaturated esters from β-keto esters with either double bond configuration.
O-Phosphorylation of chelated sodium enolates of β-keto esters with diethyl or diphenyl phosphoryl chlorides in diethyl ether give rise to the Z (trans) enol phosphate isomers
6a whereas the E (cis) isomers can be obtained through reactions with tertiary amine bases in polar solvents, eg Et
3N or
iPr
2NEt in HMPA (Scheme 2).
21,27 Vinylic couplings with methyl, primary alkyl, and
t-butyl cuprates or Grignard reagents in the presence of Cu(I) salts usually take place efficiently with retention of the double bond geometry. Palladium-catalyzed Suzuki reactions of Z and E enol triflates with methyl-,
t-butyl-, cyclopropyl-, and phenylboronic acids have been employed in similar syntheses of E and Z isoprenoid esters and their C-3 analogues.
9Scheme 2
(
E,E,
E)-Geranylgeraniol (
6) has other research applications beyond its conversion to the diphosphate. The forms labeled with deuterium and tritium at C-1 [1,1-
2H
2]- and [1-
3H]
6 can be oxidized to the corresponding labeled aldehydes which are reduced to (
R)- and (
S)-[1-
2H
1]- and [1-
3H]
6 for studies on stereochemistry of diterpene cyclase and transferase reactions.
5,28 The terminal double bonds of the benzyl ether and acetate derivatives of
6 are converted to the 14,15 bromohydrins by hypobromination reactions with
N-bromosuccinimide in aqueous THF or DME.
5,10b,29 The bromohydrins are readily cyclized to the epoxides and then converted to 14,15-oxidogeranylgeraniol.
10b Acid-catalyzed hydrolysis to the 14,15-diol and oxidative cleavage with periodate affords the trisnoraldehyde that is a key intermediate in synthesis of
6 bearing deuterium in the terminal methyl positions by E- or Z-selective olefinations and deuteride reductions.
28a Reductive amination of the trisnoraldehyde acetate and hydrolysis gives 14-aza-14,15-dihydro
6,
5 the diphosphate of which is a potent inhibitor abietadiene synthase.
30
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
(E, E)-Farnesol; (106-2805)
Methanesulfonyl chloride; (124-63-0)
Ethyl acetoacetate:
Butanoic acid,
3-oxo-,
ethyl ester; (141-97-9)
Diethyl chlorophosphate:
Phosphorochloridic acid,
diethyl ester; (814-49-3)
Diisobutylaluminum hydride:
Aluminum, hydrobis(2-methylpropyl)-; (1191-15-7)
 |
Robert M. Coates was born in Evanston, Illinois, in 1938. He received his BS degree in chemistry from Yale University in 1960 and his Ph.D degree from the University of California-Berkeley with W. G. Dauben in 1964. After postdoctoral research at Stanford University with E. E. van Tamelen in 1964–1965, he joined the faculty in the Chemistry Department at the University of Illinois at Urbana-Champaign in 1965. His research has been primarily concerned with terpenes and carbocyclic compounds, stereochemistry and mechanism of isoprenoid biosynthesis, and design and synthesis of inhibitors and active site probes for terpene synthases. |
 |
Yinghua Jin was born in Longjing, China in 1976. She received her BS degree in chemistry from Peking University in 2000. After graduation, she spent one year at University of Utah, working as a graduate assistant in the research group of Professor Frederick G. West. In 2001, she transferred to University of Illinois at Urbana-Champaign, where she completed her Ph.D degree in chemistry with Professor Robert M. Coates in 2006. Her graduate research was focused on mechanistic investigations of taxadiene biosynthesis by means of fluoro analogues of geranylgeranyl diphosphate and azanorcembrene enantiomers. |
 |
Frank G. Roberts was born in Chicago, Illinois, in 1982. He received his B.S. degree in chemistry from the University of Illinois at Urbana-Champaign in 2006 where he carried out undergraduate research on beyerane and kaurane diterpenes in R. M. Coates' laboratory. In the summer 2005 he was an exchange student at the Ludwig Maximillian University, Munich, Germany, where he worked in Prof. Hendrik Zipse's group. He is currently a Freud Fellow at the University of Chicago pursing a Ph.D degree under the direction of Professor Milan Mrksich. |
 |
Hillary M. Peltier grew up in Jeannette, Pennsylvania. She earned a B.S. from the University of Michigan, Ann Arbor in 2002. That year, she began her doctoral studies at the University of California, Berkeley in the laboratories of Prof. Jonathan A. Ellman. Her graduate work has focused on the development of methods for the asymmetric synthesis of amines using N-tert-butanesulfinamde and the application of these methods toward the synthesis of biologically relevant molecules
|
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved