Org. Synth. 2007, 84, 77
DOI: 10.15227/orgsyn.084.0077
SYNTHESIS OF TERMINAL 1,3-DIYNES VIA SONOGASHIRA COUPLING OF VINYLIDENE CHLORIDE FOLLOWED BY ELIMINATION. PREPARATION OF 1,3-DECADIYNE
Submitted by Amanda L. Kohnen and Rick L. Danheiser
1
.
Checked by Scott E. Denmark and Xiaorong Liu.
1. Procedure
A. 2-Chloro-1-decen-3-yne (2). A 350-mL, round-bottomed pressure flask (Note 1) equipped with a magnetic stir bar, rubber septum, and argon inlet needle (Note 2) is charged with (Ph3P)4Pd (3.46 g, 2.99 mmol, 0.03 equiv) (Note 3) and 150 mL of toluene (Note 4), and then vinylidene chloride (11.98 mL, 14.54 g, 150 mmol, 1.5 equiv) (Note 5) is added via syringe over 2 min. The resulting yellow mixture is stirred at room temperature for 20 min and then a mixture of 1-octyne (14.75 mL, 11.02 g, 100.0 mmol) (Note 6) and n-butylamine (14.82 mL, 10.97 g, 150 mmol, 1.5 equiv) (Note 7) is transferred into the reaction mixture via cannula over 5 min from a 50-mL, round-bottomed flask fitted with a septum and an argon inlet needle. The flask is rinsed with two 3-mL portions of toluene, which are transferred into the pressure flask by cannula. The septum is temporarily removed from the pressure flask and copper(I) iodide (0.571 g, 2.99 mmol, 0.03 equiv) (Note 8) is added in one portion. Material adhering to the sides of the pressure flask is rinsed into the reaction mixture with 5 mL of toluene. The pressure flask is then sealed under argon with a threaded Teflon cap and submerged in a preheated oil bath at 40 °C behind a safety shield. (Note 9) The color of the clear yellow solution darkens to red over 4 h. After 8 h (Note 10), the cloudy red mixture is allowed to cool to room temperature and then filtered through a medium frit sintered glass funnel to remove the precipitated ammonium salt. The solid is washed with 100 mL of hexanes, and the filtrate is concentrated by rotary evaporation (40 °C, 20 mmHg). The resulting red oil (28.0 g) is diluted with 20 mL of CH2Cl2 and concentrated onto 40 g of silica gel (40 °C, 16 mmHg, Note11). The clumpy powder is transferred to a column (25 × 8 cm) of 600 g of silica gel (Note 11) and is eluted with hexanes. After elution with 1000 mL of hexanes, 50-mL fractions are collected. Fractions 6 to 20 are combined and concentrated by rotary evaporation (40 °C, 20 mmHg) to afford 12.80–12.97 g (75–76%) of the desired enyne as a yellow liquid (Note 12).
B. 1,3-Decadiyne (3). A 1-L, three-necked, round-bottomed flask equipped with a magnetic stir bar, rubber septum, a 125-mL pressure-equalizing addition funnel fitted with a rubber septum, and a Claisen adapter fitted with an argon inlet adapter and a rubber septum penetrated by the Teflon probe of a thermometer (Notes 2, 13) is charged via syringe with diisopropylamine (23.4 mL, 16.9 g, 167 mmol, 2.6 equiv) (Note 14) followed by 200 mL of THF (Note 15). The solution is cooled at 0 °C in an ice-water bath and n-butyllithium solution (2.39 M in hexanes, 67 mL, 160 mmol, 2.5 equiv) (Note 16) is added dropwise via the addition funnel over 15 min. The resulting yellow solution is stirred at 0 °C for 30 min and then cooled to −78 °C (internal temperature) in a dry ice-acetone bath with the surface of the reaction mixture approximately 3 cm below the level of the cold acetone. A 250-mL, three-necked, round-bottomed flask equipped with a magnetic stir bar, rubber septum, glass stopper, and argon inlet adapter is charged via syringe with a solution of enynyl chloride 2 (11.0 g, 64.4 mmol, 1.0 equiv) in 80 mL of THF and cooled to −78 °C (external temperature). The solution of enynyl chloride is transferred via cannula into the solution of LDA at such a rate so as to maintain the internal temperature between −70 and −78 °C (Notes 17, 18). The 250-mL flask is washed with two 5-mL portions of THF and the washings are transferred by cannula to the flask containing the LDA. The reaction mixture, which immediately darkens to a reddish color, is allowed to stir at −78 °C for 5–10 min and then poured into a 1-L separatory funnel containing 100 mL of half-saturated NH4Cl solution. The resulting mixture is diluted with 150 mL of pentanes and extracted with 100 mL of 10% HCl solution. The organic phase is washed with 10% HCl (2 × 100 mL) and the combined aqueous phases are back-extracted with pentanes (2 × 100 mL). The combined organic layers are washed with 100 mL of saturated NaCl solution, dried over MgSO4, filtered, and concentrated by rotary evaporation (23 °C, 16 mmHg) to afford 9.40 g of a red liquid. This material is dissolved in 20 mL of CH2Cl2 and deposited onto 10 g of silica gel (Note 11) by rotary evaporation (23 °C, 16 mmHg). The resulting clumpy powder is transferred onto a column (12 × 8 cm) of 300 g of silica gel (Note 11) and is eluted with pentanes. Fractions 15 to 33 (50 mL each) are combined and concentrated under reduced pressure (23 °C, 16 mmHg) to provide 5.95–6.26 g (69–73%) of diyne 3 as an orange liquid (Note 19).
2. Notes
1.
The
350-mL, threaded, heavy-walled, round-bottomed pressure vessel (142 mm length × 82.5 mm OD) was purchased from Chemglass, Inc. The vessel was equipped with a Teflon cap (thread size 15 mm). Carrying out the reaction in a sealed vessel minimized the loss of vinylidene chloride (bp 30–32 °C).
2.
The apparatus was flame-dried under reduced pressure (0.08 mmHg) and then maintained under an atmosphere of argon during the course of the reaction.
3.
Tetrakis(triphenylphosphine)palladium(0) (99.9%) was purchased from Strem Chemicals, Inc. and used as received. The catalyst was stored at −18 °C under argon and was weighed in the glove box. The submitters observed no difference in the yield of
2 when the catalyst was stored and transferred in a dry box.
4.
Toluene was purified by pressure filtration under argon through activated alumina.
5.
Caution: Vinylidene chloride is light sensitive and highly susceptible to polymerization and peroxide formation. Vinylidene chloride (99%) was purchased from Aldrich Chemical Company, Inc. and stored in the dark at 0 °C under argon. To remove any polymers and autoxidation products, the reagent was purified immediately before use by filtration through alumina according to the following procedure. A jacketed glass column (46 cm × 2.5 cm) cooled with cold running water and equipped with a needle at the bottom was charged with 30 g of Activity I basic alumina (EM Science, 70-230 mesh). A flame-dried,
50-mL, two-necked pear-shaped flask equipped with two rubber septa was flushed with argon and attached to the needle at the bottom of the alumina column. Vinylidene chloride (approximately 25 mL) was added to the column and an exit needle was attached to the other septum of the flask. Nitrogen pressure was then applied to the top of the column to drive the vinylidene chloride through the column into the flask.
6.
1-Octyne (98%) was purchased from Alfa Aesar and used as received.
7.
n-Butylamine (99.5%) was purchased from Aldrich Chemical Company, Inc. and purified by distillation at atmospheric pressure under argon from CaH
2.
8.
Copper(I) iodide (98%) was purchased from Alfa Aesar and purified according to the following procedure. A
500-mL, round-bottomed flask was fitted with a Soxhlet extractor equipped with a reflux condenser fitted with a septum and argon inlet needle. The apparatus was flame-dried under vacuum (0.08 mmHg) and purged with argon. A Soxhlet thimble filled with
25 g of CuI was inserted into the extractor and
200 mL of THF was added via syringe. The extractor was wrapped with aluminum foil to shield the CuI from light and the THF in the flask was heated in an oil bath at reflux for 24 h. The apparatus was allowed to cool to room temperature and the CuI remaining in the thimble was transferred to a flame-dried,
100-mL, round-bottomed flask and dried at 0.08 mmHg for 24 h. The resulting CuI was ground up with a glass rod to afford a white, free-flowing powder.
9.
The checkers submerged the flask such that the surface of the solution was 1 cm below the oil bath level. However, the checkers found that the oil level had no effect on the yield so long as good stirring was maintained.
10.
The reaction was terminated when TLC analysis indicated that all of the octyne was consumed. TLC analysis was conducted using silica gel plates (elution with hexanes, visualization by UV and vanillin or KMnO
4); R
f values: octyne = 0.42, enyne product (
2) = 0.55, bis-coupling product
4 = 0.30.
11.
Silica gel was purchased from Silicycle (230-400 mesh).
12.
Enyne
2 is light sensitive and decomposes slowly at room temperature. This compound is best stored at 0 °C in the dark under argon. The product exhibits the following physical properties: R
f = 0.55 (hexanes); IR (neat) cm
−1: 2932, 2215, 1602, 1195;
1H NMR
pdf (400 MHz, CDCl
3) δ: 0.89 (t,
J = 7.1 Hz, 3 H), 1.28 – 1.33 (m, 4 H), 1.41 (app quint,
J = 7.1 Hz, 2 H), 1.55 (app quint,
J = 7.3 Hz, 2 H), 2.34 (t,
J = 7.1 Hz, 2 H), 5.51 (s, 1 H), 5.55 (s, 1 H);
13C NMR
pdf (101 MHz, CDCl
3) δ: 14.3, 19.4, 22.7, 28.3, 28.7, 31.5, 77.7, 93.0, 120.6, 121.0. Anal. Calcd. for C
10H
15Cl: C, 70.37; H, 8.86. Found: C, 70.55; H, 8.84.
13.
An Omegaette HH3087 mini-thermometer type K with a Teflon-coated thermocouple was used to monitor internal temperature.
14.
Diisopropylamine (99.5%) was purchased from Aldrich Chemical Company, Inc. and distilled at atmospheric pressure under argon from CaH
2.
15.
Tetrahydrofuran was purified by pressure filtration under argon through activated alumina.
16.
n-Butyllithium was purchased from Alfa Aesar and titrated by the method of Watson and Eastham.
217.
The position of the exit needle of the cannula was adjusted so that it is at the level of the dry ice-acetone mixture in the external cooling bath. In this fashion, the solution of enynyl chloride ran down the cold wall of the flask approximately 3 cm above the surface of the solution of LDA. If the temperature of the reaction mixture rose above −70 °C, lower yields of diyne were obtained due to isomerization (see Discussion Section).
18.
Approximately 20 min was required for the addition.
19.
1,3-Decadiyne was stored as a solution in pentanes at −20 °C under argon. The diyne exhibits the following physical properties: R
f = 0.41 (hexanes); IR (neat) cm
−1: 3310, 2957, 2298, 2226, 1466;
1H NMR
pdf (400 MHz, CDCl
3) δ: 0.88 (t,
J = 7.0 Hz, 3 H), 1.24–1.33 (m, 4 H), 1.38 (app quint,
J = 7.0 Hz, 2 H), 1.53 (app quint,
J = 7.2 Hz, 2 H), 1.96 (s, 1 H), 2.25 (t,
J = 7.0 Hz, 2 H);
13C NMR
pdf (101 MHz, CDCl
3) δ: 14.2, 19.2, 22.7, 28.2, 28.7, 31.5, 64.6, 64.8, 68.7, 78.8. Anal. Calcd. for C
10H
14: C, 89.49; H, 10.51. Found: C, 89.11; H, 10.84.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
1,3-Diynes serve as valuable building blocks for the synthesis of natural products,
3 supramolecular constructs, and a variety of oligomeric alkynes and advanced materials with interesting electronic and optical properties. The most popular approaches to the synthesis of conjugated diynes involve alkynyl coupling reactions.
4 For example, the homocoupling of acetylenes can conveniently be achieved with copper catalysis using the Glaser reaction and its various modifications While unsymmetrical 1,3-diynes are available via the Cadiot-Chodkiewicz reaction,
6 the latter method involves the copper-catalyzed coupling of an alkyne with a 1-bromoalkyne; a variant of this process employs the coupling of alkynes with 1-haloalkynes in the presence of a combination of copper and palladium catalysts.
7,8 Several strategies have been developed for the synthesis of
terminal 1,3-diynes based on the Cadiot-Chodkiewicz reaction. One approach employs the coupling of (bromoethynyl)trialkylsilanes followed by desilylation of the resultant 1-trialkylsilyl-1,3-diynes,
9 while an alternative strategy utilizes coupling reactions of propargylic alcohol derivatives followed by base-promoted fragmentation to unmask the terminal triple bond.
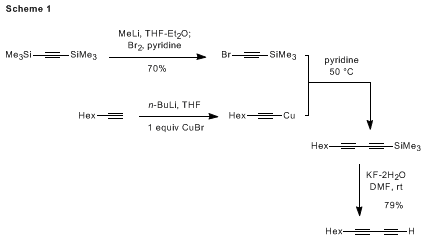
10,11 The application of the former approach to the preparation of the title compound, 1,3-decadiyne, has been reported by Zweifel
b and is outlined in Scheme 1.
12While approaches such as this provide access to a variety of internal and terminal conjugated diynes, in many cases these coupling reactions are complicated by the formation of alkyne homodimers as significant byproducts. The cost of bis(trimethylsilyl)acetylene and the need for at least three synthetic operations further limits the attractiveness of this approach as a practical method for the synthesis of terminal diynes. These limitations have motivated the development of several alternative strategies for the preparation of terminal 1,3-diynes which are based on the Sonogashira coupling
13 of an alkyne with a dihaloethylene followed by b-elimination.
The selective mono-Sonogashira coupling of 1,2-dichloroethylene with alkynes was first demonstrated by Linstrumelle in 1981,
14 and subsequently several laboratories reported the synthesis of 1,3-diynes by Sonogashira coupling of various 1,2-dihaloethylenes followed by base-promoted dehydrohalogenation.
15 The requisite 1,2-dihaloalkenes are quite expensive, however, and Negishi
16 consequently introduced a more economical approach in 2003 based on the Sonogashira coupling of inexpensive 1,1-dichloroethylene (vinylidene chloride).
17The method of Negishi provides the basis for the optimized procedure described here for the synthesis of 1,3-decadiyne. In the case of the first step, the Sonogashira coupling of vinylidene chloride and 1-octyne, the catalyst loading has been reduced relative to the original Negishi protocol (from 5 to 3 mol% each for (Ph3P)4Pd and CuI), and the amount of vinylidene chloride has been reduced from 5 to 1.5 equivalents. Our initial attempts to reduce the amount of vinylidene chloride below 5 equivalents were not successful. As the amount of vinylidene chloride was reduced, the octyne homodimer (7,9-hexadecadiyne) and enediyne 4 (the product of bis-coupling) appeared as major byproducts and the desired chloroenyne was obtained in greatly diminished yield. We hypothesized that the volatile vinylidene chloride (bp 30–32 °C) was escaping during the course of the reaction, and indeed, the formation of these byproducts could be minimized by simply carrying out the reaction in a sealed flask. Under the optimal conditions described here using 1.5 equiv of vinylidene chloride, the desired enyne was obtained in 75–76% yield and the crude coupling product was found to contain only 5–10% of enediyne 4 and less than 5% of the homodimer.
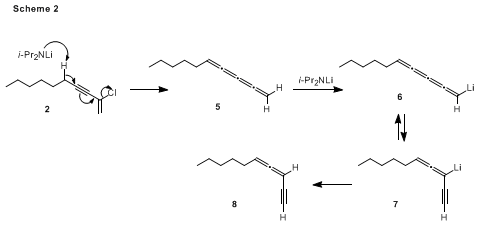
As reported by Negishi, exposure of enynyl chloride
2 to LDA at −78 °C effected β-elimination leading to the desired diyne
3. We have noted that temperature control was critical to the success of this transformation, particularly when the reaction was conducted on scales greater than 500 mg. Specifically, the allenyne
8 was formed as a byproduct in yields of up to 40% if the reaction temperature was allowed to rise above −70 °C during the addition of the chloroenyne to the solution of LDA. Scheme 2 outlines the likely mechanism for the formation of this byproduct. 1,4-Elimination
18 of
2 leads to the tetraene
5, which is further metalated by LDA to afford the organolithium intermediates
6 and/or
7. Protonation then furnishes the allenyne
8. The possibility that
8 arises from isomerization of 1,3-decadiyne was ruled out by control experiments that demonstrated that the diyne was not converted to
8 upon exposure to LDA under the conditions of the reaction.
The formation of the allenyne byproduct could be minimized by carefully controlling the temperature of the reaction mixture, especially during the addition of the chloroenyne to LDA, which resulted in an exothermic reaction. Temperature control was best achieved by precooling the solution of chloroenyne in THF to −78 °C and employing a solution of less than 1.0 M concentration. The solution was added slowly at such a rate that the internal temperature of the reaction mixture did not exceed −70 °C. Under these conditions the diyne was obtained in 66–72% yield contaminated with <2% of the allenyne.
In summary, an improved protocol for the preparation of 1,3-decadiyne (
3) by the method of Negishi has been developed. This approach employs the Sonogashira coupling of 1-octyne with vinylidene chloride followed by base-promoted dehydrochlorination with LDA. The optimized conditions described here provide 1,3-decadiyne in good yield on multigram-scale and are considerably more practical and efficient than previous methods reported for the preparation of this diyne.
19
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
n-Butylamine:
1-Butanamine; (109-73-9)
1-Octyne; (629-05-0)
Vinylidene chloride:
1,1-Dichloroethene; (75-35-4)
2-Chloro-1-decen-3-yne; (534600-74-3)
Copper(I) iodide; (7681-65-4)
1,3-Decadiyne; (55682-66-1)
Diisopropylamine:
N-(1-methylethyl)-2-propanamine; (108-18-9)
 |
Rick L. Danheiser received his undergraduate education at Columbia where he carried out research in the laboratory of Professor Gilbert Stork. He received his Ph.D. at Harvard in 1978 working under the direction of E. J. Corey on the total synthesis of gibberellic acid. Dr. Danheiser is the A. C. Cope Professor of Chemistry at MIT where his research focuses on the design and invention of new annulation and cycloaddition reactions, and their application in the total synthesis of biologically active compounds. |
 |
Amanda L. Kohnen was born in 1981 in West Hollywood and grew up in Yuma, Arizona. She did her undergraduate work at the University of California, San Diego and received a B.S. degree in Chemistry in 2004. She is now pursuing graduate studies at the Massachusetts Institute of Technology where her research under the direction of Professor Rick Danheiser involves the development of new synthetic methods and the total synthesis of biologically active natural products. Ms. Kohnen is the recipient of an MIT Leventhal Presidential Fellowship, a Pfizer Research Fellowship, and an NSF Graduate Research Fellowship. |
 |
Xiaorong Liu graduated with BS and MS degrees in Chemistry from Nanjing University in China where she carried out research on the synthesis of organic conducting polymers in the laboratory of Professor Xue Gi. In the fall of 1999, she began graduate studies under the direction of Professor Robert K. Boeckman, Jr. at the University of Rochester where her Ph.D. thesis focused on the total synthesis of (–)-Apoptolidin. In the fall of 2004, she joined the laboratory of Professor Denmark as a postdoctoral associate at University of Illinois at Urbana-Champaign. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved