Org. Synth. 2007, 84, 120
DOI: 10.15227/orgsyn.084.0120
SYNTHESIS OF 4-TRIISOPROPYLSILYL-3-BUTYN-2-OL BY ASYMMETRIC TRANSFER HYDROGENATION
[]
Submitted by James A. Marshall, Patrick Eidam, and Hilary Schenck Eidam
1a.
Checked by Kai Zhang and Dennis P. Curran
1b.
1. Procedure
A. (rac)-4-Triisopropylsilyl-3-butyn-2-ol (1). A single-necked, flame-dried, 250-mL round-bottomed flask equipped with a magnetic stir bar and a rubber septum fitted with an argon inlet needle is flushed with argon and charged with triisopropylsilyl acetylene (95%, 6.1 mL, 4.75 g, 26.0 mmol) (Note 1) and dry THF (54 mL) (Note 2) by syringe through the septum. The reaction mixture is cooled to −40 °C (bath temperature) by means of a dry ice-acetonitrile bath and tert-BuLi (1.7 M in pentane, 18 mL, 32.3 mmol) is added dropwise by means of a syringe (Note 3). The resulting bright yellow mixture is stirred at −40 °C for 30 min and then ACS reagent grade acetaldehyde (>99.5%, 2.3 mL, 39.2 mmol) is added in one portion by means of a syringe. The reaction mixture is stirred for 20 min at −40 °C and then poured over a rapidly stirring solution of saturated aqueous NH4Cl (75 mL) at room temperature. After 15 min, the phases are separated and the aqueous layer is extracted with diethyl ether (2 × 40 mL). The combined organic extracts are washed with brine, dried over MgSO4, filtered, and the solvent is removed with a rotary evaporator (40 °C, water aspirator pressure). The residue is purified by bulb-to-bulb distillation (95 °C, 0.2 mmHg; Note 4) to yield the racemic alcohol (5.8 g, 98%) as a clear oil (Notes 5, 6, 7). This material is used in the next step without further purification.
B. 4-Triisopropylsilyl-3-butyn-2-one (2). A one-necked, flame-dried, 250-mL round-bottomed flask equipped with a magnetic stir bar and a rubber septum fitted with an argon inlet needle is flushed with argon. The septum is temporarily removed and the flask is charged sequentially with CH2Cl2 (54 mL), 4-triisopropylsilyl-3-butyn-2-ol (1) (5.8 g, 25.6 mmol) and technical grade MnO2 (85%, 34.4 g, 336.9 mmol) (Notes 8, 9), which is added in one portion. The reaction mixture is stirred at room temperature for 30 min and then filtered through a short pad of Celite. The Celite is washed with CH2Cl2 (2 × 30 mL) and the solvent is removed on a rotary evaporator (40 °C, water aspirator pressure). The resulting yellow oil is purified by bulb-to-bulb distillation (80 °C, 0.2 mmHg; Note 4) to yield ketone 2 (5.0–5.1 g, 87–89%) as a clear oil (Notes 10, 11). This material is used in the next step without further purification.
C. RuCl[(S,S)-NTsCH(C6H5)CH(C6H5)NH2(η6-cymene) (3). (Note 12). A one-necked, flame-dried, 50-mL round-bottomed flask equipped with a magnetic stir bar and a rubber septum fitted with an argon inlet needle is flushed with argon. The septum is temporarily removed and the flask is charged with CH2Cl2 (6 mL), (1S,2S)-(+)-N-p-toluenesulfonyl-1,2-diphenylethylene-diamine (126 mg, 0.34 mmol), dichloro(p-cymene)ruthenium(II) dimer (107 mg, 0.17 mmol), and powdered KOH (141 mg, 2.5 mmol) (Note 1). The resulting orange mixture is stirred for 5 min at room temperature and then distilled H2O (6 mL) is added in one portion by means of a syringe. The resulting biphasic mixture is stirred for 10 min during which time the organic phase turns dark purple. The mixture is transferred to a separatory funnel, diluted with 10 mL of H2O and the layers are separated. The aqueous phase is extracted with CH2Cl2 (2 × 10 mL). The combined organic extracts are dried over CaH2 (Note 1), filtered and solvent is removed with a rotary evaporator (40 °C, water aspirator pressure) to furnish the catalyst 3 (180–182 mg, 86–87%) as a dark purple solid that is used in the subsequent reduction without additional purification.
D. (S)-4-Triisopropylsilyl-3-butyn-2-ol (4). A one-necked, flame-dried, 500-mL round-bottomed flask equipped with a magnetic stir bar and a rubber septum fitted with an argon inlet needle is flushed with argon. Isopropyl alcohol (250 mL) and 4-triisopropylsilyl-3-butyn-2-one (2) (5.1 g, 22.7 mmol) are added to the flask through the septum by syringe. The ruthenium catalyst 3 (180 mg, 0.3 mmol) is taken up in a minimal amount of CH2Cl2 (~5 mL) and added to the reaction mixture in one portion by means of a syringe. The mixture is stirred for 1.5 h and then the solvent is removed with a rotary evaporator (40 °C, water aspirator pressure). The brown residue is purified by bulb-to-bulb distillation (95 °C, 0.2 mmHg; Note 4) to yield the alcohol 4 (5.0 g, 85% for three steps) as a clear oil (Notes 13, 14, 15).
2. Notes
1.
All chemicals were purchased from Aldrich Chemical Co. and were used without further purification. The submitters purchased
THF and
CH2Cl2 as optidry Fischer Paks from Fischer Scientific. The checkers purchased
dry CH2Cl2 from Aldrich and used THF from a sodium/benzophenone still.
2.
The submitters reported that the use of diethyl ether in place of THF resulted in diminished yields and the formation of impurities not separable by distillation.
3.
The submitters reported that the use of
n-BuLi (20.2 mL of 1.6 M
n-BuLi in hexanes) afforded the adduct
1 in quantitative yield. The spectra of the sample prepared in this way were identical to those of the sample from the
tert-BuLi experiment.
4.
Bulb-to-bulb distillations were performed with an Aldrich Kugelrohr apparatus.
5.
Physical characteristics for 4-triisopropylsilyl-3-butyn-2-ol
(1): R
f = 0.41 (15% EtOAc-hexane) on Merck silica gel 60 F 254 precoated 250 μm plates; IR (film): 3325, 2943, 2866, 2173, 1464 cm
−1;
1H NMR
pdf (300 MHz, CDCl
3) δ: 1.08 (m, 21 H), 1.48 (d,
J = 6.6 Hz, 3 H), 1.78 (s, 1 H), 4.55 (q,
J = 6.6 Hz, 1 H);
13C NMR
pdf (75 MHz, CDCl
3) δ: 11.1, 18.5, 24.5, 58.7, 84.3, 109.8.
6.
The submitters reported that the product was contaminated by up to 3% triisopropylacetylene in some runs. This was analyzed by integration of the alkynyl H signal at 2.35 ppm in the
1H NMR spectrum.
7.
An analytical sample was prepared by chromatography on silica gel. A 1.5 cm × 20 cm glass column packed with 9 g of EMD silica gel 60 was used to purify 20 mg of alcohol by elution with 10% diethyl ether in hexanes. Anal. Calcd for C
13H
26OSi: C, 68.96; H, 11.57. Found: C, 68.96; H, 11.66.
8.
MnO2 was purchased from Aldrich Chemicals and dried in an oven at 110 °C overnight prior to use.
9.
The submitters reported that exposure to less than 13 equiv of MnO
2 resulted in incomplete conversion and formation of byproducts upon prolonged reaction times.
10.
Physical characteristics for 4-triisopropylsilyl-3-butyn-2-one
(2): R
f = 0.76 (15% EtOAc-hexane) on Merck silica gel 60 F 254 precoated 250 μm plates; IR (film): 2946, 2868, 2147, 1684, 1464, 1196 cm
−1;
1H NMR
pdf (300 MHz, CDCl
3) δ: 1.10 (m, 21 H), 2.37 (s, 3 H);
13C NMR
pdf (75 MHz, CDCl
3) δ: 11.3, 18.4, 32.8, 95.2, 104.6, 184.4.
11.
An analytical sample was prepared by chromatography on silica gel. (A 1.5 cm × 20 cm glass column packed with 9 g of EMD silica gel 60 was used to purify 20 mg of ketone by elution with 5% diethyl ether in hexanes. Anal. Calcd for C
13H
24OSi: C, 69.58; H, 10.78. Found: C, 69.56; H. 10.89.
12.
The procedure described for the preparation of RuCl[(
S,
S)-NTsCH(C
6H
5)CH(C
6H
5)NH
2(η
6-cymene) was taken from reference 9.
13.
Physical characteristics for
(S)-4-triisopropylsilyl-3-butyn-2-ol (
4): Same as for racemic 4-triisopropylsilyl-3-butyn-2-ol; [α]
D 20 −21.3 (c = 1.58, CHCl
3). The er of this material was found to be >95:5 by
19F NMR analysis [δ−73.68 ppm (major) and −73.02 ppm (minor), in CDCl
3 versus benzotrifluoride as an internal standard at −63.72 ppm] of the
(R)-Mosher ester. To prepare the Mosher ester, 4-triisopropylsilyl-3-butyn-2-ol (4 mg),
(S)-(+)-methoxy-a-trifluoromethylphenylacetyl chloride (5 μL) and
dry pyridine (0.15 mL) were added to a
one-necked, flame-dried, 5-mL round-bottomed flask. The flask was equipped with a magnetic stir bar and a rubber septum and was flushed with argon. The reaction mixture was stirred at room temperature for 2 h, then the volatiles were evaporated to yield the crude (
R)-Mosher ester, which was directly analyzed by
19F NMR spectroscopy without purification.
14.
GC analysis on a β-Dex column temperature programmed from 130 to 180 °C gave a single peak at 22.35 min (submitters). GC analysis on an Agilent MP-1 column (1909/Z-413E, 30 m × 0.32 mm), temperature ramp 10 °C per min from 50 °C to 315 °C, gave a single peak at 10.61 min (checkers). Anal. Calcd for C
13H
26OSi: C, 68.96; H, 11.57. Found: C, 69.03; H, 11.74.
15.
The submitters measured [α]
D 20 −19.5 (c = 1.55, CHCl
3) for a run on the same scale and concentration. When they performed the reduction at a ten-fold higher concentration of ketone, the derived alcohol
4 exhibited [α]
D 20 −16.5 (c = 1.81, CHCl
3).
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
The palladium-catalyzed conversion of enantioenriched propargylic mesylates such as
2 to enantioenriched allenylzinc (
3) or indium (
4) reagents and in situ additions to aldehydes provides a convenient route to homopropargylic alcohol intermediates related to polyketide natural products (Scheme 1).
2,3,4Scheme 1. Synthesis of Enantioenriched Homopropargylic Alcohols from Propargylic Mesylates
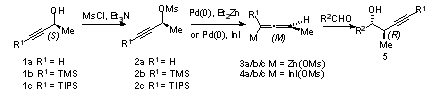
Although the alcohol precursor of mesylate (
S)-
2a and its enantiomer are commercially available,
5 their high cost discourages widespread use of the methodology. Accordingly, we have explored simple and economical preparations through use of chiral pool starting materials
6 or lipase resolution of racemic propargylic alcohols for a more cost effective synthesis.
7,8 The TMS reagents
2b, prepared through lipase resolution of racemic
1b, show significantly higher diastereoselectivity than the parent alkyne
2a. While this alternative represents an improvement, the intrinsic inefficiency of optical resolution coupled with the volatility of the resolved alcohols represent significant drawbacks to the methodology. The present procedure employs Noyori asymmetric hydrogenation of alkynyl ketones to prepare the alkynyl TIPS derivative
1c of 3-butyn-2-ol.
9 This route offers several major advantages compared with the previous approaches to the parent
1a and the TMS alcohol
1b:
- Unlike alcohol 1a and its alkynyl TMS analog 1b, the TIPS compound 1c is nonvolatile and completely insoluble in water, thereby allowing facile isolation by simple extraction;
- all intermediates can be vacuum-dried and purified by short path distillation;
- both enantiomers of the chiral catalyst are readily available;
- the catalyst precursor can be stored without loss of activity; and,
- addition reactions of the derived TIPS allenylzinc reagent to aliphatic aldehydes proceeds readily and with comparable diastereoselectivity to additions of the parent reagent or the TMS derivative.
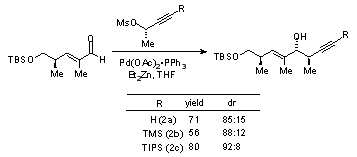
The outcome of addition reactions of the three allenylzinc reagents generated in situ from mesylates 2a, 2b, and 2c to cyclohexane-carboxaldehyde is summarized in Table 1.
Table 1. Comparison of Addition Reactions of Allenylzinc Reagents Derived in situ from the Mesylate of (S)-3-Butyn-2-ol (2a) and the Silylated Analogs 2b and 2c to Cyclohexanecarboxaldehyde.
An even larger difference in diastereoselectivity for the three reagents was observed with an α-methylated enal (Table 2). Removal of the TIPS group from the adducts is easily effected in minutes with tetrabutylammonium fluoride in THF.
Table 2. Comparison of Addition Reactions of Allenylzinc Reagents Derived in situ from the Mesylate of (S)-3-Butyn-2-ol (2a) and the Silylated Analogs 2b and 2c to a Conjugated Aldehyde.
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
(rac)-4-Triisopropylsilyl-3-butyn-2-ol:
3-Butyn-2-ol, 4-[tris(1-methylethyl)silyl]-: (726202-65-9)
Triisopropylsilyl acetylene:
Silane,
ethynyltris(1-methylethyl)-; (89343-06-6)
tert-BuLi:
Lithium,
(1,1-dimethylethyl)-; (594-19-4)
Acetaldehyde; (75-07-0)
4-Triisopropylsilyl-3-butyn-2-one:
3-Butyn-2-one,
4-[tris(1-methylethyl)silyl]-: (183852-48-4)
Manganese Dioxide; (1313-13-9)
RuCl[(S,S)-NTsCH(C6H5)CH(C6H5)NH2(η6-cymene):
Ruthenium,
[N-[(1S,2S)-2-(amino-κN)-1,2-diphenylethyl]-4-methyl-benzenesulfonamidato-κN]chloro[(1,2,3,4,5,6-η)-1-methyl-4-(1-methylethyl)benzene]-; (192139-90-5)
(1S,2S)-(+)-N-p-toluenesulfonyl-1,2-diphenylethylenediamine:
Benzenesulfonamide,
N,N'-(1,2-diphenyl-1,2-ethanediyl)bis[4-methyl-,
[S-(R*,R*)]-; (170709-41-8)
Dichloro(p-cymene)ruthenium(II) dimer; (52462-29-0)
(S)-4-Triisopropylsilyl-3-butyn-2-ol:
(S)-3-Butyn-2-ol, 4-[tris(1-methylethyl)silyl]-;
 |
James Marshall received a BS degree in Chemistry from the University of Wisconsin PhD degree in 1960 from the University of Michigan with Professor Robert E. Ireland. After two years as an NIH postdoctoral Fellow in the laboratory of William. S. Johnson at Stanford University, he joined the faculty of Northwestern University in 1962 and advanced to the rank of full Professor in 1968. The following year he moved to the University of South Carolina and was subsequently appointed the inaugural Guy Lipscomb Professor of Chemistry. In 1995 he was appointed Thomas Jefferson Professor of Chemistry at the University of Virginia. A central theme in Professor Marshall's research has been the development and application of stereoselective reactions to natural product synthesis. |
 |
Patrick M. Eidam was born in 1978 in New Castle, PA. Following his graduation with a BS degree in Chemistry from Lafayette College, he entered graduate school at the University of Virginia and joined the research group of Professor James Marshall. He received his PhD degree in 2006 and subsequently obtained a position with the Medicinal Chemistry division at Glaxo Smith Kline in King of Prussia, PA. |
 |
Hilary Schenck Eidam was born in 1978 in York, PA. She received a B.S. degree in chemistry from Gettysburg College in 2000 followed by a PhD degree in chemistry from the University of Virginia in the laboratory of Milton Brown. After postdoctoral research with Professor James Marshall at Virginia she joined the Medicinal Chemistry group at Glaxo Smith Kline in King of Prussia, PA.
|
 |
Kai Zhang was born in China and got his B.S. degree in Chemistry from University of Science and Technology of China in 2005. He came to United States in August 2005 as a graduate student and joined Professor Dennis P. Curran's group at the University of Pittsburgh. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved