Org. Synth. 2007, 84, 163
DOI: 10.15227/orgsyn.084.0163
PREPARATION OF A TRISAMIDOMOLYBDENUM(VI) PROPYLIDYNE COMPLEX
[− A HIGHLY ACTIVE CATALYST PRECURSOR FOR ALKYNE METATHESIS]
Submitted by Wei Zhang, Yunyi Lu, and Jeffrey S. Moore
1.
Checked by Günter Seidel and Alois Fürstner.
1. Procedure
CAUTION: Procedures involving molybdenum trisamide 4 were conducted under an atmosphere of argon. It has been reported that 4 is able to activate molecular nitrogen at or below room temperature.2 Therefore, N2 must not be used as a protecting atmosphere for any experiment involving this reagent (Note 1).
A. N-(3,5-Dimethylphenyl)-tert-butylamine (1). A 500-mL, three-necked, round-bottomed flask is equipped with two glass stoppers, a magnetic stir bar and a reflux condenser fitted with a gas inlet connected to an argon-vacuum manifold. The flask is flame-dried under vacuum, then filled with argon. After evacuating and backfilling with argon three times, the flask, in which a positive flow of argon is maintained throughout the entire procedure, is sequentially charged with sodium tert-butoxide (19.22 g, 0.20 mol) (Note 2), 2-(dicyclohexylphosphino)biphenyl (0.50 g, 1.4 mmol), [Pd2(dba)3] (0.65 g, 0.71 mmol) (Note 3), toluene (200 mL) (Note 4), tert-butylamine (18.0 mL, 0.17 mol) and 5-bromo-1,3-dimethylbenzene (19.4 mL, 0.14 mol) (Note 2). The resulting viscous red reaction mixture is heated at 80 °C in an oil bath for 12 h while maintaining positive argon pressure. The solution color changes from dark red to orange red with white solids precipitating out. The mixture is transferred to a 500-mL, one-necked, round-bottomed flask and concentrated to a residue by rotary evaporation (water bath temperature 50–55 °C) before cooling down to room temperature (Note 5). tert-Butyl methyl ether (250 mL) (Note 2) and brine (80 mL) are added to the residue. After separation, the aqueous layer is extracted with tert-butyl methyl ether (2 × 250 mL), the combined organic layers are dried (MgSO4), filtered and concentrated by rotary evaporation, and the crude product is purified by distillation under vacuum (bp 50–53 °C, 10−3 mbar, oil bath temperature 70 °C) affording aniline 1 as colorless liquid (21.8 g, 86%) (Note 6).
B. Lithium N-(3,5-dimethylphenyl)-tert-butylamide etherate (2). A 500-mL, three-necked, round-bottomed flask is equipped with a magnetic stirring bar, two glass stoppers and a pressure-equalizing dropping funnel fitted with a gas inlet connected to an argon-vacuum manifold. The flask is flame-dried under vacuum, then filled with argon. After evacuating and backfilling with argon three times (Note 1), the flask is charged with substituted aniline 1 (11.3 g, 63.7 mmol) and n-hexane (220 mL) (Note 4) under argon purge. The solution is cooled to −78 °C in a dry ice-acetone bath and a solution of n-BuLi (1.6 M in hexanes, 48 mL, 76.8 mmol) (Note 2) is added via the dropping funnel over 10 min. After the addition is complete, the mixture is stirred for 12 h during which time it is gradually warmed to room temperature; a white precipitate forms upon warming. The dropping funnel is replaced by a distillation head and condenser and all volatiles are removed under vacuum (approximately 7.5 mmHg) at room temperature without exposing the contents to the open atmosphere (Notes 1, 7). Then ether (25 mL) and n-hexane (50 mL) (Note 4) are added to the remaining viscous liquid causing further precipitation of a white solid. The resulting suspension is kept at −30 °C under argon for 12 h (Note 1, 7). The white solids of lithium anilide etherate (2•Et2O) formed are collected by filtration under argon, washed with cold n-hexane (2 × 10 mL) and dried under vacuum (10−3 mbar) to give lithium N-(3,5-dimethylphenyl)-tert-butylamide etherate as colorless crystals (9.41 g, 57%). The filtrate is concentrated under vacuum to approximately half of its volume and slowly cooled to −78 °C to give a second crop of the product which is isolated by filtration under argon as described above (3.48 g, 21%) (Note 8).
C. [MoCl3(THF)3](3). A 500-mL, two-necked, round-bottomed flask equipped with a magnetic stir bar, a glass stopper and a gas inlet connected to the argon/vacuum line is flame-dried and back-filled with argon. While maintaining a positive argon pressure, the flask is charged with acetonitrile (250 mL) (Note 4). Molybdenum (V) chloride (MoCl5) (51.17 g, 160 mmol) is then added in portions over a period of 20 min, causing a gentle warming of the mixture. The resulting suspension is stirred for 20 h at ambient temperature. The solid material is filtered off and is carefully rinsed with acetonitrile (50 mL) before it is dried under vacuum (10−3 mbar) to give MoCl4(MeCN)2 as a red-brown solid (47.5 g, 93%). This material is re-dispersed in THF (200 mL) and the suspension is stirred for 3.5 h under argon. The resulting complex is filtered off under argon, the solid is carefully washed with pentane (100 mL) and dried under vacuum (10−3 mbar) to give MoCl4(THF)2 as orange-brownish crystals (53.30 g, 94%).
A 500-mL, two-necked, round-bottomed flask equipped with a magnetic stir bar, a glass stopper and a gas inlet connected to the argon/vacuum line is flame-dried and back-filled with argon. This flask is charged with the MoCl4(THF)2 complex (53.30 g, 140 mmol), THF (300 mL) and tin shot (107.3 g, 0.904 mol), and the resulting suspension is stirred for 30 min, resulting in a slight exotherm (a temperature of approximately 30 °C is reached). Stirring is then discontinued to allow the remaining tin to settle. Upon gentle manual swirling, leaving the metallic tin at the bottom of the flask, the supernatant suspension (liquid and all solids except metallic tin) is decanted, the solid material is collected by filtration under argon, the product is carefully rinsed with pentane (100 mL) and dried under vacuum (10−3 mbar), affording MoCl3(THF)3 as an orange solid (51.2 g, 88%) (Note 9).
D. Tris[N-(tert-butyl)(3,5-dimethylphenyl)-amido]molybdenum(III) (4). A 500-mL two-necked, round-bottomed flask equipped with a magnetic stir bar, a glass stopper and a gas inlet connected to the argon/vacuum manifold is flame-dried under vacuum and back-filled with argon (Note 1). The flask is then charged with [MoCl3(THF)3] 3 (7.96 g, 19.0 mmol) by removing a stopper and adding the solid, followed by Et2O (300 mL) (Note 4), which is transferred by syringe. The resulting suspension is cooled to −30 °C with an acetone/dry ice bath before anilide 2 (9.79 g, 38.0 mmol) is added through removal of the stopper. The resulting mixture is stirred for 3 h during which time it is allowed to reach room temperature and the solution turns dark red. The suspension is filtered under argon into a flame-dried 500-mL, two-necked, round-bottomed flask to remove LiCl; the solid is rinsed with ether (15 mL). With one neck equipped with a rubber septum and the other one connected via a cold-trap to the vacuum line, the filtrate is concentrated under reduced pressure (7.5 mmHg) to approximately 1/6 of its original volume (50 mL) (Note 10). The remaining solution is cooled to - 30 °C for 16 h, causing the separation of dark red crystals. The supernatant liquid is decanted off while keeping a positive argon pressure, and the remaining dark-red, crystalline molybdenum trisamide 4 is dried under vacuum (10−3 mbar) (4.28 g, 55%) (Note 11).
E. Tris[N-(tert-butyl)(3,5-dimethylphenyl)-amido]molybdenum(VI) propylidyne (5). A 250-mL, two-necked, round-bottomed flask equipped with a rubber septum, a magnetic stirring bar, and a gas inlet connected to the argon/vacuum manifold is flame-dried and back-filled with argon (Note 1, 10). Molybdenum trisamide 4 (5.31 g, 8.48 mmol), magnesium turnings (2.54 g, 106 mmol) (Note 2) and THF (160 mL) (Note 4) are added to the flask by removal of the septum. To the stirred solution, 1,1-dichloropropane (1.7 mL, 17 mmol) (Note 2, Note 12) is added dropwise via syringe within 5 min, resulting in a color change from red to dark amber. The resulting mixture is stirred for 2.5 h at room temperature under argon. The rubber septum is replaced by a distillation head connected to the vacuum line, the solvent is removed under reduced pressure (approximately 7.5 mmHg) (Note 10), and the residue is redissolved in pentane (100 mL) (Note 4). After vigorous stirring/manual shaking, the solid precipitate (MgCl2) and excess magnesium turnings are removed by filtration under argon. The filtrate is concentrated to approximately 1/10 of its volume and slowly cooled to −78 °C, causing the molybdenum propylidyne complex to precipitate (Note 13). The supernatant is removed via cannula and the remaining beige crystals are dried under vacuum (10−3 mbar) (3.08 g, 54%) (Notes 1, 14, 15).
2. Notes
1.
All compounds described in this procedure, except
N-(3,5-dimethylphenyl)-
tert-butylamine (
1), are highly sensitive to air and moisture. Therefore, proper protection from atmospheric influences must be maintained throughout the preparation and handling of these products. The checkers ensured this by applying Schlenk techniques, whereas the submitters largely relied on the use of an argon-filled glove box.
2.
Sodium tert-butoxide (97%),
tert-butylamine (98%),
5-bromo-1,3-dimethylbenzene (97%),
tert-butyl methyl ether (≥ 99%),
n-butyllithium (1.6 M in hexanes),
magnesium turnings (~3/16 in., 99.98%),
1,1-dichloropropane (97%, dried using activated, powdered 3Å molecular sieves overnight before use),
p-nitrophenol (≥ 99%),
1-phenyl-1-butyne (99%, passed through a short plug of basic alumina and transferred under vacuum before use) were purchased from Aldrich Chemical Company, Inc. and used as received unless further purification indicated.
3.
2-(Dicyclohexylphosphino)biphenyl (98%),
[Pd2(dba)3], MoCl5 (99.6%) and
tin shot (99.8%) were purchased from Strem Chemicals, Inc. and used as received.
4.
The checkers used solvents purchased from Acros which were purified and deoxygenated by distillation under argon over the drying agents indicated: toluene, hexane, pentane (Na/K alloy), Et
2O, THF,
tert-butyl methyl ether (Mg-anthracene), MeCN (CaH
2). The submitters used toluene, hexane, Et
2O, THF and pentane deoxygenated by bubbling argon through them for a period of 20 min and dried using 3 Å molecular sieves overnight before use.
5.
When cooling down to room temperature, the reaction mixture turned into a viscous solution, making the direct solvent removal under vacuum impractical. In case this happens,
tert-butyl methyl ether (250 mL) and
H2O (100 mL) should be added, and the aqueous layer removed. The organic layer should be concentrated under vacuum and the residue subjected to further workup as described in the text.
6.
The procedure for synthesizing aniline
1 follows that reported by Fürstner, et al.
3a The spectral characteristics of compound
1 are as follows:
1H NMR
pdf (CDCl
3, 400 MHz) δ: 1.31 (s, 9 H), 2.22 (s, 6 H), 3.30 (s, 1 H), 6.38 (s, 2 H), 6.41 (s, 1 H).
13C NMR
pdf (CDCl
3, 100 MHz) δ: 21.5, 30.1, 51.3, 115.4, 120.2, 138.3, 146.8. According to
1H NMR and GC/MS, the product contains approximately 3% of 5-bromo-1,3-dimethylbenzene as an impurity that cannot be separated at this stage. The spectroscopic data of
1 are in agreement with those reported in the literature.
3a7.
With identical results, the reaction can be set up in the glove box.
8.
The procedure for synthesizing anilide
2 follows that reported by Fürstner.
3a The spectral characteristics of compound
2 are as follows:
1H NMR
pdf (300 MHz,
d8-toluene) δ: 0.91 (t,
J = 6.8 Hz, 6 H), 1.57 (s, 9 H), 2.29 (s, 6 H), 3.13 (q,
J = 6.8 Hz, 4 H), 6.11 (s, 1 H), 6.52 (s, 2 H). These data are in agreement with those reported in the literature.
3a9.
MoCl
3(THF)
3 prepared according to this procedure shows the following analytical properties: Elemental analysis calcd. for C
12H
24Cl
3MoO
3 (418.62): C 34.43, H 5.78; found: C 34.54, H 5.59. The submitters reported a "one-pot" preparation of
3 following a procedure reported by Poli.
4 Under these conditions, however, the checkers got erratic results with the appearance of the samples obtained in different runs ranging in color from orange to brown to lilac; in none of the cases was the elemental analysis correct. Therefore, the stepwise procedure outlined in the text is highly recommended.
10.
The submitters performed this procedure in a glove box. The vacuum source was obtained by connecting an oil pump and a liquid nitrogen cold trap to a gas inlet with a hose in the glove box. The flask can be refilled with argon by closing the vacuum line and punching the septum with a needle. During concentration under vacuum, the flask was gently heated with a heat gun occasionally to speed the evaporation.
11.
The procedure for synthesizing molybdenum triamide
4 follows that reported by Fürstner.
3a The spectral characteristics of compound
4 are as follows:
1H NMR
pdf (300 MHz,
d8-toluene) δ: −50.4 (br. s, 3 H), −20 (br. s, 6 H), −9.3 (br. s, 18 H), 61.7 (br, 27 H). Trisamide
4 can be used as such as an efficient alkyne metathesis precatalyst upon
in situ activation with CH
2Cl
2 in toluene.
3a,9,1012.
Trisamide
4 was treated with 2 equiv of 1,1-dichloropropane instead of higher equiv in order to minimize a side product that presumably arises from a Grignard reaction.
13.
The title compound's spectral characteristics are as follows:
1H NMR
pdf (300 MHz, C
6D
6) δ: 1.45 (br. s, 30 H), 2.12 (s, 18 H), 3.13–3.95 (m, 2 H), 6.05 (br. s, 6 H), 6.66 (s, 3 H);
13C NMR
pdf (150 MHz, THF-
d8) δ: 14.3 (br), 21.7, 33.9 (br), 45.6 (br), 61.2 (br), 127.7 (br), 130.9 (br), 137.3, 151.6 (br), 303.2; MS (EI, vaporization temperature = 130 °C):
m/z (%): 665 (76) [M
+], 610 (100), 552 (80), 490 (41), 432 (32), 391 (44), 375 (70), 335 (48), 306 (19); HRMS (EI) (C
39H
59MoN
3): calcd for
98Mo: 667.3764, found 667.3761. The deuterated solvents used were dried and deoxygenated by distillation over potassium metal chips before use. The product contained small amounts (< 5 wt% based on
1H NMR integration) of
N-
tert-butyl aniline
1,
7 which could not be removed even upon repeated recrystallization from pentane. Therefore, the product was used for alkyne metathesis without any further purification.
14.
The submitters report testing of the metathesis activity of catalyst precursor
5 as follows: In the glove box under an atmosphere of argon
(Note 1), toluene-
d8 (0.7 mL) was added to a mixture of
molybdenum(VI) propylidyne 5 (3.7 mg, 0.0056 mmol) and
p-nitrophenol (2.3 mg, 0.017 mmol) (Note 2) in a sealable NMR tube
(Note 16). The color of the resulting catalyst solution changed from yellow to dark red within 4 min of mixing
(Note 17). Then
1-phenyl-1-butyne (6) (16.0 μL, 0.112 mmol) (Note 2) was added to the catalyst solution. After mixing well, the metathesis reaction was monitored in this sealed NMR tube at 20 °C. The
1H NMR (d1 = 5 s)
(Note 18) spectrum was recorded until the ratio of 3-hexyne to
6 was constant. The reaction reached equilibrium within 10 min with 46% conversion obtained under closed system conditions
(Note 19).
15.
The submitters reported isolation of the
molybdenum propylidyne complex 5 (5.05 g, 89%) as a light yellow powder by simple evaporation of the pentane under vacuum in the glove box. The product can be further purified by recrystallization from pentanes as originally reported.
5 The submitters did not observe noticeable difference in the catalytic activity of the catalyst generated from
5 after recrystallization or from
5 without recrystallization.
16.
J. Young valve NMR tube (Wilmad-Labglass, product No. 528-JY-8) was used.
17.
A red precipitate was observed during mixing, which was presumably due to the low solubility of the generated species in toluene. In the case of chloroform, ethyl acetate or THF as solvent, no precipitation was observed.
18.
A long relaxation time (d1 = 5 s) was employed to ensure the accurate
1H NMR integration on the signals of both starting compound
6 and products.
19.
Ferrocene was used as internal standard and the mass balance was higher than 95%. Conversion was calculated from the integrated intensities of product and starting material. The equilibrium position was established by conducting the metathesis reaction in both forward (starting from
6) and backward (starting from 3-hexyne and diphenylacetylene) directions. In both cases, the same molar ratio (1.0:1.7) of dimer to monomer was obtained.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Alkyne metathesis has attracted great interest in the past two decades due to its powerful application in both organic synthesis and polymer chemistry.
9 Ring closing alkyne metathesis (RCAM) has been successfully applied to natural products synthesis,
10 and acyclic diyne metathesis (ADIMET) has been employed in preparation of high molecular weight polymers.
11 More recently, one-step gram-scale synthesis of shape-persistent arylene ethynylene macrocycles from simple monomer precursors have also been accomplished.
8The application of alkyne metathesis depends on the availability and performance of the alkyne metathesis catalysts, especially with regard to convenience of catalyst synthesis, substrate compatibility, and temperature required for catalytic activity. The most readily available of these catalysts is a poorly characterized species.
12 Many of the known catalysts require high temperature (≥ 130 °C) for metathesis activity, and exhibit limited functional group tolerance. The first well-defined alkyne metathesis catalyst was the tungsten alkylidyne complex (
t-BuO)
3W=C(
t-Bu) developed by Schrock in the early 1980's.
13 Recently, Cummins has reported a new route to synthesize analogous trialkoxymolybdenum(VI) alkylidyne complexes (R
3O)
3Mo=CR
1, which are active alkyne metathesis catalysts even at room temperature.
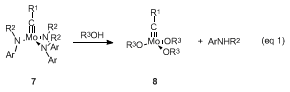
14 Although trialkoxy alkylidyne
8 can be conveniently obtained from alcoholysis of the metathesis inactive triamide
7 (eq 1), the starting complex
7 15 requires a time-consuming, multistep synthetic procedure and presents a practical limitation to this potentially useful catalyst.
A related alkyne metathesis catalyst was developed by Fürstner who combined molybdenum trisamide
4 with methylene chloride to produce a mixture of ClMo[N(
t-Bu)Ar]
3 (
9) and HC=Mo[N(
t-Bu)Ar]
3 (
10) (eq 2). This mixture exhibited alkyne metathesis activity at 80 °C with good functional group compatibility.
3a,b The catalytically active species is derived from monochloride
9, whereas methylidyne
10 exhibits poor metathesis activity.
16Given that Fürstner's method directly produces alkylidyne 10, which is the precursor to Cummins' active trialkoxyalkylidyne 8, an expedient synthesis of complex 7 is envisioned to involve redirecting the reaction in eq 2 to favor production of HC=Mo[N(t-Bu)Ar]3. Magnesium selectively reacts with monochloride 9 returning it to starting complex 4, while complex 10 remains unaffected. In the presence of excess methylene chloride, the net result is a reductive recycle approach to selectively generate the desired complex 10 in one pot (eq 3).
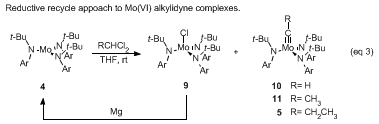
Using this method, trisamidomolybdenum(VI) ethylidyne
11 and −propylidyne
5 were prepared in high yield by the treatment of trisamide
4 with 1,1-dichloroethane or 1,1-dichloropropane, respectively, in the presence of magnesium. Treatment of trisamidomolybdenum(VI) propylidyne (
5) − the catalyst precursor − with various phenols/alcohols produced alkyne metathesis catalysts active at room temperature. The Mo(VI) catalyst with
p-nitrophenol as the ligand showed the highest catalytic activity.
17 In contrast to the reported incompatibilities of the tungsten alkylidyne complex (
t-BuO)
3W=C(
t-Bu) with amines or polyether chains,
3a,10 the molybdenum(VI) propylidyne catalyst prepared through the reductive recycle strategy displayed good compatibility with these and other functional groups, as well as a wide variety of solvents.
17 This catalyst has been successfully used in synthesis of small molecules,
17 defect-free poly(thienyleneethynylene)s (PTEs) (
12) and poly(phenyleneethynylene)s PPEs (
13) with high molecular weight.
18 A variety of arylene ethynylene macrocycles (
14,
15) have also been successfully prepared in one step starting from monomers catalyzed by this Mo(VI) propylidyne catalyst.
8,19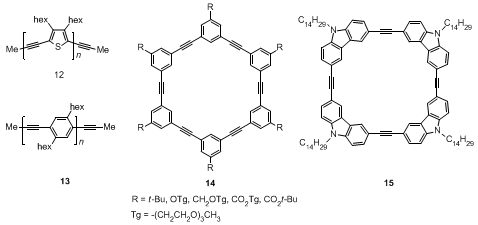
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
Sodium tert-butoxide:
2-Propanol, 2-methyl-, sodium salt; (865-48-5)
2-(Dicyclohexylphosphino)biphenyl:
Phosphine, [1,1'-biphenyl]-2-yldicyclohexyl-; (247940-06-3)
Tris(dibenzylideneacetone)dipalladium(0) [Pd2(dba)3]:
Palladium, tris[μ-[(1,2-η:4,5-η)-(1E,4E)-1,5-diphenyl-1,4-pentadien-3-one]]di-; (52409-22-0)
tert-Butylamine:
2-Propanamine, 2-methyl-; (75-64-9)
5-Bromo-m-xylene:
Benzene, 1-bromo-3,5-dimethyl-; (556-96-7)
N-(3,5-Dimethylphenyl)-tert-butylamine:
Benzenamine, N-(1,1-dimethylethyl)-3,5-dimethyl-; (110993-40-3)
Butyllithium; (109-72-8)
Lithium N-(3,5-dimethylphenyl)-tert-butylamide etherate:
Lithium, [N-(3,5-dimethylphenyl)tricyclo[3.3.1.13,7]decan-1-aminato][1,1'-oxybis[ethane]]-; (215180-13-5)
Molybdenum(V) chloride; (10241-05-1)
Tris[N-(tert-butyl)(3,5-dimethylphenyl)-amido]molybdenum(III): (236740-70-8)
1,1-Dichloropropane; (78-99-9)
Trisamidomolybdenum(VI) propylidyne:
Molybdenum, tris[N-(1,1-dimethylethyl)-3,5-dimethylbenzenaminato]propylidyne-, (T-4)-; (616886-28-3)
p-Nitrophenol:
4-Nitrophenol; (100-02-7)
1-Phenyl-1-butyne:
1-Butynylbenzene; (622-76-4)
 |
Jeffrey Moore was born near Joliet, IL in 1962. He received his BS in chemistry (1984) from the University of Illinois, and his PhD in Materials Science and Engineering with Samuel Stupp (1989). Following a NSF postdoctoral fellowship at Caltech with Robert Grubbs and an Assistant Professor appointment at the University of Michigan in Ann Arbor, he returned in 1993 to the University of Illinois, where he is currently the William H. and Janet G. Lycan Professor of Chemistry and Materials Science and Engineering. His research focuses on molecular self-assembly, structure-controlled macromolecules and foldamers, stimuli-responsive materials, and self-healing polymers.
|
 |
Wei Zhang was born in China in 1977. After completing his BS in chemistry in 2000 at Peking University and one year graduate study in University of Utah, he transferred to the University of Illinois at Urbana-Champaign in 2001, where he completed his PhD in chemistry with Prof. Jeffrey Moore, working on the development of molybdenum-based alkyne metathesis catalysts and their application to the synthesis of shape-persistent macrocycles and conjugated polymers (2005). In March 2006 he began a postdoctoral position at MIT with Prof. Timothy M. Swager.
|
 |
Yunyi Lu was born in China in 1980. She received her BS in Chemistry from Nanjing University in 2003. In the same year, she began her graduate studies at the University of Illinois with Professor Jeffrey Moore. Currently she is working on the preparation and study of polymer nanofiltration membranes for water purification.
|
 |
Günter Seidel (born 1947) studied Chemistry at the Fachhochschule Aachen Jülich to become an academically trained engineer. He is working at Max-Planck-Institute for Coal Research in Mülheim/Ruhr, Germany, since 1972. Until 1992 he worked on the preparation of recent organic boron compounds and their application in organic synthesis. Since 1993 he is working in Prof. Fürstner's group in the field of organometallic chemistry, C-C coupling reactions, transition-metal catalysts, and their application in the alkene- and alkyne metathesis. He is co-author of 83 publications and 1 patent.
|
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved