Org. Synth. 2007, 84, 272
DOI: 10.15227/orgsyn.084.0272
PREPARATION OF BENZOCYCLOBUTENONE DERIVATIVES BASED ON AN EFFICIENT GENERATION OF BENZYNES
[Bicyclo[4.2.0]octa-1,3,5-trien-7-one, 5-(phenylmethoxy)-]
Submitted by Shin-ichiro Tsujiyama and Keisuke Suzuki
1a
.
Checked by David B. Guthrie, Heather M. Gibney, and Dennis P. Curran
1b.
1. Procedure
Caution! Iodine, trifluoromethanesulfonic anhydride, benzyl bromide and hydrofluoric acid are toxic and/or corrosive reagents and should be handled in a well-ventilated hood. Calcium gluconate gel should be present for first aid in case of accidental exposure of skin to HF.
A. 2-Iodoresorcinol (1) (Note 1). A 250-mL, one-necked, round-bottomed flask equipped with a large, elliptical stir bar and open to the atmosphere is charged with distilled water (46 mL), resorcinol (7.27 g, 66.0 mmol) and iodine (17.92 g, 70.6 mmol), and then placed in an ice-water bath (Note 2). To this open flask is slowly added sodium bicarbonate (6.16 g, 73.3 mmol) in portions by spatula over 5 min at 0 °C with vigorous stirring (Notes 2, 3). During the addition, vigorous gas evolution (carbon dioxide) is observed. The ice bath is removed and the mixture is allowed to warm to room temperature over 20 min, then stirred for a further 10 min, during which time it becomes a brown slurry (Note 4). The products are extracted with ethyl acetate (3 × 50 mL). The combined organic extract is successively washed with 10% aqueous sodium thiosulfate solution and brine, dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure (Note 5). The solid residue is triturated in chloroform (20 mL) for 10 min at –10 °C (ice/ethanol bath), filtered, and washed with cold (–10 °C) chloroform (20 mL) to give 9.12 g (58%) of 2-iodoresorcinol (1) as a cream-colored solid. The filtrate is concentrated under reduced pressure, and the residue is triturated in chloroform (7 mL) for 30 min at –10 °C, filtered, and washed with 5 mL of chloroform (0 °C) to give an additional 1.18 g (8%, 66% combined yield) of 2-iodoresorcinol (1) as a cream-colored solid. (Notes 6, 7, 8, 9).
B. 3-Benzyloxy-2-iodophenyl trifluoromethanesulfonate (2). An oven-dried, 500-mL, one-necked, round-bottomed flask equipped with a magnetic stirring bar, an argon gas inlet and a rubber septum is charged with 2-iodoresorcinol (1) (11.49 g, 48.7 mmol) and dichloromethane (115 mL) (Note 10), and the mixture is cooled to –78 °C with a dry ice-acetone bath. N,N-Diisopropylethylamine (20.4 mL, 117 mmol) is added by syringe. Then trifluoromethanesulfonic anhydride (18.0 mL, 107 mmol) is slowly added over 15 min by syringe pump while maintaining the reaction mixture at –78 °C (Note 11). After stirring for 10 min at –78 °C, the reaction mixture is warmed to 0 °C by replacing the dry ice bath with an ice water bath, and stirred for 1 h (Note 12). The reaction is quenched by the slow addition of water (100 mL, added over 10 min), and the products are extracted with dichloromethane (2 × 50 mL). The combined organic extract is successively washed with saturated aqueous sodium bicarbonate solution (2 × 50 mL) and brine (2 × 50 mL), dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The residue is dissolved in diethyl ether (50 mL) and hexanes (50 mL), and then silica gel (12.0 g) (Note 13) is added. After stirring for 30 min at room temperature, the mixture is filtered, and the silica cake is washed with a mixture of diethyl ether (75 mL) and hexanes (75 mL). The filtrate is concentrated under reduced pressure and vacuum dried to give 24.31–24.33 g of crude 2-iodoresorcinol bis(trifluoromethanesulfonate) as a dark brown oil. This is used directly for the next step without further purification.
A 500-mL, one-necked round-bottomed flask equipped with a magnetic stirring bar and a reflux condenser attached to an inert atmosphere inlet is placed under argon and charged with the crude 2-iodoresorcinol bis(trifluoromethanesulfonate) (24.3 g), 1,2-dimethoxyethane (100 mL) and cesium carbonate (19.06 g, 58.5 mmol) (Note 14). The resulting mixture is stirred for 4 h in an 80 °C oil bath (Note 15). After cooling to room temperature, benzyl bromide (6.96 mL, 58.5 mmol) (Note 16) is added via syringe over 2 min, then the reaction mixture is stirred for 5 h at room temperature (Note 17). Diethylamine (2.5 mL, 24.4 mmol) (Note 18) is added via syringe through the top of the condenser, and the mixture is stirred for 1 h at room temperature. The mixture is cooled in an ice bath and 1 M hydrochloric acid (200 mL) is added, dropwise at first, then more rapidly as the exotherm subsides. The mixture is warmed to room temperature and the products are extracted with ethyl acetate (3 × 100 mL). The combined organic extract is successively washed with brine (2 × 100 mL), saturated aqueous sodium bicarbonate (2 × 100 mL), and brine (2 × 100 mL). The extracts are dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure to give 26.3–26.5 g of a brown oil. The residue is purified by column chromatography on silica gel (Note 19) to give 16.5–17.4 g (74–78%) of 3-benzyloxy-2-iodophenyl trifluoromethane- sulfonate (2) as yellow crystals (Note 20), which are used directly for the next step.
C. 5-Benzyloxybicyclo[4.2.0]octa-1,3,5-trien-7-one (3). An oven-dried, single-necked 500-mL round-bottomed flask equipped with a magnetic stirring bar, an argon inlet and a rubber septum is charged with 3-benzyloxy-2-iodophenyl trifluoromethanesulfonate (2) (12.6 g, 27.5 mmol). The flask is flushed with argon, tetrahydrofuran (76 mL) (Note 21) and 1-(tert-butyldimethylsilyloxy)-1-methoxyethene (8.99 mL, 41.2 mmol) (Note 21) are added by syringe, and the mixture is cooled to –78 °C (bath temperature) with a dry ice-acetone bath. A solution of butyllithium in hexanes (20.63 mL of a 1.6 M solution, 33.0 mmol) (Note 22) is added to the reaction mixture over 20 min via syringe pump using a needle inserted through the septum. The reaction mixture is further stirred for 10 min at –78 °C (Note 23), then water (2 mL) is added dropwise. After warming to room temperature, water (100 mL) is added, and the products are extracted with ethyl acetate (3 × 100 mL). The combined organic extract is successively washed with brine (3 × 100 mL), dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure to give 10.5–10.8 g of crude 5-benzyloxy-7-tert-butyldimethylsilyloxy-7-methoxy-bicyclo- [4.2.0]octa-1,3,5-triene as a brown oil. This is used directly for the next step without further purification.
A 250-mL, three-necked, round-bottomed flask (Note 24) equipped with a magnetic stirring bar, an addition funnel, a stopper and an argon inlet is charged with 5-benzyloxy- 7-tert-butyldimethylsilyloxy-7- methoxybicyclo[4.2.0]octa-1,3,5-triene (10.5–10.8 g), acetonitrile (82 mL), and the mixture is cooled to 0 °C in an ice-water bath. Aqueous hydrofluoric acid (48%, 12.5 mL, 345 mmol) (Note 25) is slowly added during 2 min via addition funnel. The mixture is warmed to room temperature over 20 min, then stirred an additional 20 min (Note 26). The reaction mixture is carefully poured into saturated aqueous sodium bicarbonate solution (300 mL) (Note 27), and the products are extracted with ethyl acetate (3 × 100 mL). The combined organic extract is successively washed with saturated aqueous sodium bicarbonate solution (3 × 100 mL) and brine (3 × 100 mL), dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure to give 6.24–7.08 g of a yellow solid. The solid is taken up in hot hexanes (40 mL warmed to 60 °C) and the resulting mixture is quickly filtered to remove small amounts of insoluble brown solid. Cooling to 0 °C in an ice bath followed by filtration of the precipitate and washing with cold hexanes gives 3.72–4.34 g (60–70%) of 5-benzyloxybicyclo[4.2.0]octa-1,3,5-trien-7-one (3) as a white solid (Notes 28, 29).
2. Notes
1.
The iodination process was based on the procedure by Thomsen, I.; Torssell, K. B. G.
Acta Chem. Scand.
1991,
45, 539.
2.
The submitters used distilled
water, iodine and sodium bicarbonate purchased from Kanto Chemical Co., Inc. Resorcinol was purchased from Tokyo Kasei Kogyo Co., Inc. The checkers used Aldrich HPLC grade distilled water.
Sodium bicarbonate was purchased from EMD,
resorcinol from Lancaster (99%), and
iodine from Aldrich or Fisher (ACS reagent grade, >99.8%). All reagents were used without further purification.
3.
The checkers used an elliptical stir bar shaped like an American football, 32 mm long and 17 mm in diameter at the widest point. The submitters used a mechanical stirrer. Rapid stirring was essential for the success of the reaction, and a good vortex was required to prevent a foam from forming on the surface of the mixture during the addition of bicarbonate. When the stirring was not efficient, less 2-iodoresorcinol was formed along with increased amounts of 4-iodoresorcinol, 2,4-diiodoresorcinol and unreacted resorcinol. Sodium bicarbonate was added every few seconds in portions of about 55–65 mg by spatula.
4.
Reaction progress was monitored by TLC analysis on Merck silica gel 60 F254 plates, visualized by a 254-nm UV lamp and stained with an aqueous solution of phosphomolybdic acid. TLC analysis showed the formation of 2-iodoresorcinol (
1) (3/2 hexanes/ethyl acetate, R
f = 0.48). The side products (4-iodoresorcinol and 2,4-diidoresorcinol) had similar R
f values to the target product, while the R
f of resorcinol was 0.28.
5.
The residue usually solidified spontaneously at this point. If not,
chloroform (20 mL) was added and the mixture was again evaporated.
6.
The submitters obtained a first crop from trituration of 9.49 g (61%) and a second crop of
2.18 g (14%) for a total yield of 2-iodoresorcinol (1) of 75%.
7.
A reaction conducted at twice the indicated scale gave
21.86 g (70%) 2-iodoresorcinol (
1) after two titrations (20.11 g, then 1.75 g).
8.
The product was stored in a dark bottle at 5 °C because slow decomposition occurred on exposure to ambient light at room temperature.
9.
Mp 99–101 °C;
1H NMR
pdf (300 MHz, acetone-
d6) δ: 6.46 (d,
J = 8.1 Hz, 2 H), 7.00 (t,
J = 8.1 Hz, 1 H), 8.83 (s, 2 H);
13C NMR
pdf (75 MHz, acetone-
d6) δ: 75.4, 107.1, 130.4, 158.8; IR (thin film, CHCl
3): 3456, 3363 (broad), 1593, 1582, 1491, 1458, 1304, 1278, 1250, 1185, 1159, 1022, 994, 784, 704 cm
−1; EIMS (
m/z): M
+ 237 (6), 236 (100), 218 (23), 127 (16), 81 (13), 63 (8), 53 (17); HRMS (
m/z): [M]
+ calcd for C
6H
5IO
2, 235.9334; found, 235.9333. Anal. calcd. for C
6H
5IO
2: C, 30.54; H, 2.14. Found: C, 30.27; H, 2.14.
10.
Dichloromethane, purchased from Asahi Glass Co., Inc. or Aldrich Chemical Co, was distilled from phosphorus pentoxide and calcium hydride (submitters) or dried by passing through an activated alumina column (checkers).
11.
N,N-Diisopropylethylamine was purchased by the submitters from Tokyo Kasei Kogyo Co., Inc., and
trifluoromethanesulfonic anhydride was donated from Central Glass Co., Inc. These reagents were used without further purification. The submitters purchased these chemicals from Aldrich Chemical Co. and redistilled the amine before use.
12.
TLC analysis showed the formation of 2-iodoresorcinol bis(trifluoromethanesulfonate) (4/1 hexanes/ethyl acetate, R
f = 0.57).
13.
Silica gel (60 Å, for column chromatography) was purchased from Kanto Chemical Co., Inc. (submitters) or Sorbent Technologies (60 Å, 230 × 400 mesh) (checkers).
14.
1,2-Dimethoxyethane was purchased from Tokyo Kasei Kogyo Co., Inc. (submitters) or J. T. Baker (checkers).
Cesium carbonate (Reagent Plus, 99%) was purchased from Aldrich Chemical Co. These reagents were used without further purification.
15.
TLC analysis showed the formation of 3-hydroxy-2-iodophenyl trifluoromethanesulfonate (4/1 hexanes/ethyl acetate, R
f = 0.24).
16.
Benzyl bromide purchased from Tokyo Kasei Kogyo Co., Inc. (submitters) or Aldrich Chemical Co. (Reagent grade, 98%) (checkers) was used without further purification.
17.
TLC analysis showed the formation of 3-benzyloxy-2-iodophenyl trifluoromethanesulfonate (
2) (4/1 hexanes/ethyl acetate, R
f = 0.45).
18.
Diethylamine was used to quench the excess benzyl bromide.
Diethylamine was purchased from Kanto Chemical Co., Inc. (submitters) or Aldrich Chemical Co. (Reagent Plus, >99.5%) (checkers) and was used without further purification.
19.
The product was taken up in 3/1 hexanes/ethyl acetate and stirred for 1 h. The organic solution was decanted from a small amount of dark brown solid directly onto the column. The chromatography was performed on 300 g of silica gel eluting with 10/1 hexanes/ethyl acetate.
20.
Mp 86–87 °C;
1H NMR
pdf (300 MHz, CDCl
3) δ: 5.21 (s, 2 H), 6.86 (d,
J = 8.1 Hz, 1 H), 6.98 (d,
J = 8.4 Hz, 1 H), 7.32–7.40 (m, 2 H), 7.40–7.48 (m, 2 H), 7.48–7.53 (m, 2 H);
13C NMR
pdf (75 MHz, CDCl
3) δ: 71.4, 83.3, 111.6, 114.3, 118.7 (q,
JC-F = 318.8 Hz), 126.9, 128.1, 128.6, 130.3, 135.6, 151.2, 159.4; IR (thin film, CHCl
3): 1589, 1450, 1425, 1273, 1224, 1209, 1139, 1056, 1024, 947, 850, 826, 732 cm
−1; EIMS (
m/z): M
+ 459 (6), 458 (36), 382 (24), 368 (11), 367 (7), 249 (11), 197 (52), 107 (64), 92 (98), 79 (44), 69 (78), 65 (100); HRMS (
m/z): [M]
+ calcd for C
14H
10F
3IO
4S, 457.9297; found, 457.9316. Anal. calcd. for C
14H
10F
3IO
4S: C, 36.70; H, 2.20. Found: C, 36.77; H, 2.19.
21.
1-(tert-Butyldimethylsilyloxy)-1-methoxyethene was obtained from Aldrich Chemical Co., Inc., and used without further purification. The submitters observed that distillation of 1-(
tert-butyldimethylsilyloxy)- 1-methoxyethene under argon did not result in increased product yield or purity. The submitters used
tetrahydrofuran (dehydrated, stabilizer free) purchased from Tokyo Kasei Kogyo Co., Inc. without further purification. The checkers used
THF from Aldrich Chemical Co. (99%, inhibitor free), which was dried by passing through activated alumina.
22.
Butyllithium in hexanes was purchased from Kanto Chemical Co., Inc. (submitters) or Aldrich Chemical Co. (checkers).
23.
TLC analysis showed consumption of starting material and formation of 5-benzyloxy-7-
tert-butyldimethylsilyloxy-7-methoxybicyclo- [4.2.0]octa-1,3,5-triene (4/1 hexanes/ethyl acetate, R
f = 0.74).
24.
The submitters and checkers used a glass vessel for this reaction, but the submitters reported that a polypropylene flask could also be used to avoid corrosion of the flask by hydrofluoric acid.
25.
The submitters used acetonitrile and a 46% aqueous solution of
hydrofluoric acid, purchased from Kanto Chemical Co., Inc. without further purification. The checkers used acetonitrile from J. T. Baker and 48% HF from Mallinckrodt.
26.
TLC analysis showed the formation of 5-benzyloxybicyclo- [4.2.0]octa-1,3,5-trien-7-one (
3) (4/1 hexanes/ethyl acetate: R
f = 0.55).
27.
Vigorous evolution of carbon dioxide gas was observed.
28.
Mp 80–81 °C;
1H NMR
pdf (300 MHz, CDCl
3) δ: 3.95 (s, 2 H); 5.47 (s, 2 H), 6.90 (d,
J = 8.4 Hz, 1 H), 7.06 (d,
J = 6.9 Hz, 1 H), 7.30–7.43 (m, 3 H), 7.43–7.51 (m, 3 H);
13C NMR
pdf (75 MHz, CDCl
3) δ: 51.1, 73.8, 115.1, 116.3, 127.7, 128.0, 128.3, 132.4, 136.3, 137.6, 150.4, 152.1, 184.7; IR (thin film, CHCl
3): 3033, 2929, 1761, 1604, 1573, 71475, 1453, 1386, 1274, 1160, 1129, 1052, 787, 755 cm
−1; EIMS (
m/z): M
+ 224 (20), 105 (6), 91 (100), 65 (13), 51 (14). HRMS (
m/z): [M]
+ calcd for C
15H
12O
2: 224.0837; found: 224.0845. Anal. calcd. for C
15H
12O
2: C, 80.34; H, 5.39. Found: C, 80.36; H, 5.10.
29.
The submitters obtained a second crop as follows. The filtrate was concentrated under reduced pressure to give 1.62 g of a yellowish solid, which was recrystallized from hexanes (6 mL, at 60 °C followed by cooling down to 0 °C using an ice bath) to give the second crop of
3 (0.60 g, 10%). In the checkers' hands, the second crop product was about 90–95% pure according to
1H NMR spectroscopy.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Benzocyclobutenes have long attracted much attention due to their molecular structure, chemical reactivity and synthetic potential.
2 They have served as important building blocks for syntheses of various natural products by exploiting their unique reactivity. Due to their inherent strain, benzocyclobutenes are capable of isomerizing to quinodimethanes that can in turn be used to derive various polycyclic skeletons via Diels-Alder reactions or electrocyclizations. Described here is an effective procedure for the synthesis of highly oxygenated benzocyclobutenones, which have great synthetic potential, since a carbonyl group in the four-membered ring can serve as a platform to introduce various functionalities. The present protocol is particularly attractive because it provides benzocyclobutene analogues in assorted oxidation states by the use of various ketene silyl acetals (Scheme 1).
3 These products can be important synthetic intermediates for various polyaromatic natural products, such as gilvocarcin,
4 pradimicin,
5 aquayamycin,
6 and TAN-1085.
7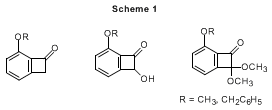
Arynes are important reactive intermediates, and many studies of their generation and reactions have been undertaken.
8 Several protocols for generation of arynes have been known basically relying on the 1,2-elimination on aromatic rings. These include treatment of aryl halides with a strong base such as sodium amide,
8 decomposition of benzenediazonium-2-carboxylate,
9 halogen-metal exchange on
ortho-dihalosubstituted benzenes,
10 and treatment of
ortho-trimethylsilylphenyl triflates with fluoride.
11 However, all methods require high reaction temperatures and/or long reaction times, and often accompanied by side reactions.
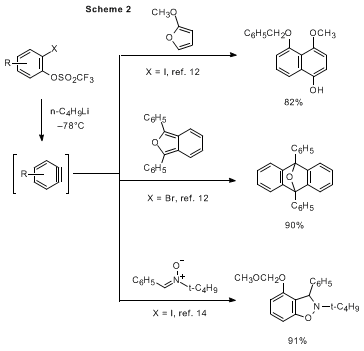
In the present procedure, an
ortho-iodoaryl triflate is employed as an efficient benzyne precursor.
12 This is easily prepared by mono-cleavage of a
bis-triflate by cesium carbonate
13 and in situ-benzylation of the resulting cesium phenoxide. Benzynes are immediately generated by treatment of the
ortho-iodoaryl triflate with butyllithium at low temperature (typically –78 °C). Compared with former methods, the present protocol enables faster generation of benzynes at much lower temperature because of the rapid halogen-lithium exchange and the ability of the triflate to serve as an excellent leaving group. The benzyne intermediate thus generated combines with arynophiles with high-lying HOMOs such as ketene silyl acetals,
3 furans,
12 and nitrones
14 in reactions that afford the corresponding cycloadducts in high yields (Scheme 2).
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
Resorcinol:
1,3-Benzenediol (9); (108-46-3)
Iodine:
HIGHLY TOXIC. CORROSIVE. (9); (7553-56-2)
2-Iodoresorcinol:
1,3-Benzenediol, 2-iodo- (9); (41046-67-7)
N,N-Diisopropylethylamine:
Triethylamine, 1,1'-dimethyl- (8); 2-Propanamine, N-ethyl-N-(1-methylethyl)- (9); (7087-68-5)
Trifluoromethanesulfonic anhydride:
CORROSIVE:
Methanesulfonic acid, trifluoro-, anhydride (9); (358-23-6)
2-Iodoresorcinol bis(trifluoromethanesulfonate):
Methanesulfonic acid, trifluoro-, 2-iodo-1,3-phenylene ester (9); (514826-78-9)
Cesium carbonate (7);
Carbonic acid, dicesium salt (9); (534-17-8)
3-Hydroxy-2-iodophenyl trifluoromethanesulfonate:
Methanesulfonic acid, trifluoro-, 3-hydroxy-2-iodophenyl ester (9); (514826-79-0)
Benzyl bromide:
CORROSIVE. LACHRYMATOR:
Toluene, α-bromo- (8);
Benzene, (bromomethyl)- (9); (100-39-0)
Diethylamine (8); Ethanamine, N-ethyl- (9); (109-89-7)
3-Benzyloxy-2-iodophenyl trifluoromethanesulfonate:
Methanesulfonic acid, trifluoro-, 2-iodo-3-(phenylmethoxy)phenyl ester (9); (138720-02-2)
1-(tert-Butyldimethylsilyloxy)-1-methoxyethene:
Silane, (1,1-dimethylethyl)[(1-methoxyethenyl)oxy]dimethyl- (9); (77086-38-5)
n-Butyllithium:
FLAMMABLE LIQUID:
Lithium, butyl- (9); (109-72-8)
Hydrofluoric acid:
CORROSIVE. (9); (7664-39-3)
5-Benzyloxybicyclo[4.2.0]octa-1,3,5-trien-7-one:
Bicyclo[4.2.0]octa-1,3,5-trien-7-one, 5-(phenylmethoxy)- (9); (169615-68-3)
 |
Keisuke Suzuki was born in 1954. He received his Ph.D. in 1983 from the University of Tokyo under Professor Teruaki Mukaiyama. He began his academic career at Keio University in 1983, and moved to the Tokyo Institute of Technology in 1996. He was the recipient of the Japan Chemical Society Award for Young Chemists in 1986, Japan IBM Award in 1994, Nagoya Silver Medal in 1999, and Synthetic Organic Chemistry Award, Japan in 2003. His research interests include the exploitation of new synthetic methods for selective synthesis of natural products.
|
 |
Shin-ichiro Tsujiyama was born in Nagasaki, Japan in 1974. After undergraduate studies at Gifu Pharmaceutical University, he joined the group of Prof. T. Kataoka, and obtained his M.Sc. degree in pharmacology in 1999. Currently he is working for Fujifilm Finechemicals Co., Ltd. in process chemistry, and also is a Ph.D. program student in the group of Prof. K. Suzuki at Tokyo Institute of Technology on studies of benzynes and nitrile oxides.
|
 |
David B. Guthrie received his B.S. degree at Saint Vincent College of Latrobe, PA in 2003 and performed research with Dr. Daryle Fish. He is currently pursuing his Ph.D. degree at the University of Pittsburgh under the supervision of Prof. Dennis P. Curran. His doctoral studies have included mechanistic investigations of 5-endo-trig radical cyclizations, and chiral transfer in reactions of axially chiral o-halo anilides.
|
 |
Heather Gibney was born in 1982 in Pennsylvania, USA. She graduated from Juniata College with a B.S. degree in chemistry where she worked with Professor Richard R. Hark synthesizing tripeptides using solid phase chemistry. She then attended the University of Pittsburgh and in 2006 received her M.S. degree under Professor Dennis P. Curran where she studied radical atom transfer cyclizations. She is now a coatings chemist at Sauereisen, Inc. in Pittsburgh, PA, where she formulates corrosion resistant coatings and technical cements.
|
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved