Org. Synth. 2007, 84, 295
DOI: 10.15227/orgsyn.084.0295
PROTECTION OF ALCOHOLS USING 2-BENZYLOXY-1-METHYLPYRIDINIUM TRIFLUOROMETHANESULFONATE: METHYL (R)-(-)-3-BENZYLOXY-2-METHYL PROPANOATE
Submitted by Kevin W. C. Poon, Philip A. Albiniak, and Gregory B. Dudley
1.
Checked by Martin A. Berliner and John A. Ragan
2.
1. Procedure
Caution: Exercise care in the handling of 18-Crown-6 (harmful, target organ: nerves) and methyl triflate (corrosive, causes burns, target organs: eyes, skin, mucous membranes).
A. 2-Benzyloxypyridine. A 500-mL, three-necked, round-bottomed flask equipped with a magnetic stir bar, a thermocouple probe, a glass stopper, and a Dean-Stark trap with reflux condenser and a nitrogen bubbler (Note 1) is charged with benzyl alcohol (11.7 g, 0.108 mol, 1.0 equiv), 2-chloropyridine (13.5 g, 0.119 mol, 1.1 equiv) (Note 2), potassium hydroxide (20.0 g, 0.356 mol, 3.3 equiv) (Note 3), and toluene (210 mL) (Note 4) by temporary removal of the glass stopper. The suspension is stirred and 18-crown-6 (1.43 g, 5.40 mmol, 0.05 equiv) (Note 2) is added in one portion. The reaction mixture is heated to reflux (Note 5) with azeotropic removal of water (Note 6) for 1 h. The reaction mixture is then allowed to cool to 25 °C. The fittings of the flask are removed. Water (100 mL) is added to the cooled reaction mixture and the resulting biphasic solution is transferred to a 500-mL separatory funnel. The reaction flask is rinsed with 50 mL of toluene and the rinse is combined with the reaction mixture. The lower, strongly basic aqueous layer (pH 14) is removed. The organic phase is washed a second time with water (100 mL) and the aqueous layer (pH ~7) is removed. The cloudy, colorless organic phase is transferred to a 500-mL flask and concentrated under reduced pressure on a rotary evaporator (25–45 mmHg, bath temperature 45–50 °C). Residual water remaining in the organic phase is azeotropically removed during the concentration, to provide the crude product as a clear and colorless to pale yellow oil. This material is transferred to a 100-mL single-necked round-bottomed flask with magnetic stir bar and purified by vacuum distillation using a short-path distillation apparatus. A single fraction is collected (bp 93–95 °C, 1.0 mmHg) providing 19.0–19.2 g (95–96%) of 2-benzyloxypyridine as a colorless liquid (Notes 7, 8).
B. 2-Benzyloxy-1-methylpyridinium trifluoromethanesulfonate. A three-necked, 250-mL, round-bottomed flask equipped with a nitrogen bubbler inlet (Note 1), thermocouple probe, rubber septum and overhead mechanical stirrer (Note 9) is charged with 2-benzyloxypyridine (10.6 g, 57.3 mmol, 1.0 equiv) and toluene (60 mL) (Note 4) by temporary removal of the septum. The resulting solution is cooled to 0 °C in an ice bath. Methyl trifluoromethanesulfonate (6.80 mL, 60.1 mmol, 1.05 equiv) (Note 10) is added dropwise via syringe over 15 min, during which time white solids crystallize and the internal temperature increases to approximately 15 °C. The ice bath is removed and the viscous slurry is allowed to warm to ambient temperature. After 1 h (Note 11), the heterogeneous reaction mixture is diluted with 50 mL of hexanes (Note 12) and the solids are isolated by filtration and washed with one additional portion of hexanes (50 mL). After drying under vacuum, 19.6–19.8 g (98.5–99%) of 2-benzyloxy-1-methylpyridinium trifluoromethanesulfonate is isolated as a white, microcrystalline solid (Note 13).
C. Methyl (R)-(-)-3-benzyloxy-2-methyl propanoate. An oven-dried, 250-mL, three-necked, round-bottomed flask is equipped with a reflux condenser with an inert gas bubbler, a magnetic stir bar, a thermocouple probe and a glass stopper. 2-Benzyloxy-1-methylpyridinium trifluoromethanesulfonate (16.7 g, 48.0 mmol, 2.0 equiv) (Note 14), magnesium oxide (1.94 g, 48.0 mmol, 2.0 equiv) (Note 15), α,α,α-trifluorotoluene (48 mL) (Note 16) and methyl (R)-(-)-3-hydroxy-2-methyl propionate (2.65 mL, 23.8 mmol, 1.0 equiv) (Note 17) are charged to the reaction flask by temporarily removing the glass stopper. The heterogeneous reaction mixture is then heated in an oil bath (83 °C bath temperature, 80–82 °C internal temperature) for 24 h (Note 18). The reaction mixture is allowed to cool to room temperature and suction filtered through a pad of Celite on a sintered glass funnel. The Celite is washed with methylene chloride (2 × 20 mL). The filtrate is concentrated by rotary evaporation (15 mmHg, 25 °C bath temperature), and the light brown oil is purified by flash column chromatography (Note 19) to provide 3.77–4.10 g (76–82%) of methyl (R)-(-)-3-benzyloxy-2-methyl propanoate as a colorless liquid (Notes 20, 21).
2. Notes
1.
The submitters conducted this procedure under an atmosphere of argon; the checkers found substitution of nitrogen to have no adverse impact on yield or purity.
2.
Benzyl alcohol (99%),
2-chloropyridine (99%), and
18-crown-6 (99%) were purchased from Sigma-Aldrich and used without further purification.
3.
Potassium hydroxide pellets were purchased from J. T. Baker and ground with a mortar and pestle immediately before use.
4.
The submitters purchased
toluene from VWR and passed it through neutral alumina and 4Å molecular sieves columns under argon atmosphere before use. The checkers obtained ACS grade toluene from Sigma-Aldrich and used it as received.
5.
The checkers utilized a heating mantle and a solid state heating controller set to 125 °C at approximately 80% power to maintain reflux. The submitters utilized an oil bath (150 °C bath temperature).
6.
At the end of the reaction, 1.4 mL of water was collected in the Dean-Stark trap.
7.
The checkers performed a simple distillation under vacuum with the distillation pot heated in an oil bath set at 120 °C. The submitters employed fractional vacuum distillation using a short-path distillation apparatus fitted with a 3-neck pig adapter and thermometer and connected to a vacuum oil pump (0.25 mmHg).
8.
The compound had the following physical data: bp: 93–95 °C/ 1.0 mmHg. IR (thin film): 1594, 1569, 1472, 1430, 1270, 988, 777, 733, 695 cm
−1.
1H NMR
pdf (400 MHz, CDCl
3) δ: 5.36 (s, 2 H), 6.80 (dt,
J = 8.4, 0.9 Hz, 1 H), 6.87 (ddd,
J = 7.1, 5.1, 0.9 Hz, 1 H), 7.30 (m, 1 H), 7.34–7.39 (m, 2 H), 7.44–7.49 (m, 2 H), 7.57 (ddd,
J = 8.4, 7.1, 2.0 Hz, 1 H), 8.17 (ddd,
J = 5.1, 2.0, 0.8 Hz, 1 H);
13C NMR
pdf (100 MHz, CDCl
3) δ: 67.5, 111.3, 116.9, 127.8, 127.9, 128.4, 137.3, 138.6, 146.8, 163.6; Anal: Calcd for C
12H
11NO: C, 77.81; H, 5.99; N, 7.56. Found: C, 77.56; H, 6.07; N, 7.32. R
f = 0.64 (250 μm silica gel on glass, hexanes:ethyl acetate = 3:1, UV visualization). The spectral data for 2-benzyloxypyridine matched that of the literature data.
39.
The submitters used magnetic stirring for this reaction. The checkers observed that cooling of the thick slurry was much less efficient with magnetic stirring than with mechanical stirring at the scale of this preparation.
10.
Methyl trifluoromethanesulfonate (MeOTf) was purchased from Sigma-Aldrich, stored and handled under an atmosphere of argon, and used without further purification.
11.
TLC analysis showed disappearance of 2-benzyloxypyridine within 1 h by TLC (R
f (reaction product) = 0; R
f (starting material) = 0.6; 3:1 hexanes:EtOAc; UV visualization) and by HPLC (product t
R 2.7 min; starting material t
R 2.4 min; Agilent Zorbax SB-CN column, 4.2 × 75 mm, 3.5 μ silica; mobile phases 0.1% HClO
4/H
2O and acetonitrile; isocratic 95:5 aqueous:acetonitrile for 0.25 min, ramp to 10:90 over 5 min; isocratic at 10:90 for 0.75 min, flow 2 mL/min, UV detection at 210 nm, 30 °C column temperature).
12.
The checkers obtained ACS grade hexanes from Sigma-Aldrich and used it as received.
13.
Further purification of the material thus obtained was not necessary. The compound had the following physical data: mp: 92–93 °C. IR (thin film): 1639, 1587, 1518, 1254, 1148, 1028, 772, 751, 699, 633 cm
−1.
1H NMR
pdf (400 MHz, CDCl
3) δ: 4.07 (s, 3 H), 5.54 (s, 2 H), 7.38–7.52 (m, 6 H), 7.60 (d,
J = 8.8 Hz, 1 H), 8.31 (ddd,
J = 1.8, 7.5, 9.2 Hz, 1 H), 8.47 (dd,
J = 6.4, 1.5 Hz, 1 H);
13C NMR
pdf (100 MHz, CDCl
3) δ: 41.8, 74.3, 112.0, 119.0, 120.6 (q,
JCF = 320 Hz), 128.5, 128.9, 129.5, 132.5, 143.7, 148.0, 159.4;
19F NMR
pdf (376 MHz, CDCl
3, external reference to TFA) δ: −78.8 (s); MS (APCI+) 200.2 (M-OTf)
+. Anal: Calcd for C
14H
14F
3NO
4S: C, 48.14; H, 4.04; N, 4.01; Found: C, 48.27; H, 3.73; N, 3.95. The submitters also reported the following data: HRMS (ESI+) found 200.1070 (M-OTf)
+ (calcd for C
13H
14NO
+: 200.1075) CHFNOS analysis: Calcd for C
14H
14F
3NO
4S: C, 48.14%; H, 4.04%; F, 16.31%; N, 4.01%; O, 18.32%; S, 9.18%. Found: C, 48.24%; H, 4.09%; F, 16.27%; N, 4.10%; O, 18.53%; S, 9.31%.
14.
A minimum of 1.8 equiv of 2-benzyloxy-1-methylpyridinium trifluoromethanesulfonate was found to be necessary for complete consumption of the alcohol substrates. Dibenzyl ether (Bn
2O) was formed as a by-product during the course of the reaction, perhaps through reaction of the pyridinium salt with MgO and/or adventitious moisture to generate benzyl alcohol
in situ.
15.
The checkers obtained magnesium oxide (light) from Sigma-Aldrich and dried it in a vacuum oven at 60 °C for 12 h. The submitters dried magnesium oxide under vacuum in a flask immersed in an oil bath for 6 h and stored under an atmosphere of argon.
16.
α,α,α-Trifluorotoluene in a Sure/Seal bottle was purchased from Sigma-Aldrich and used without further purification.
17.
Methyl (R)-(-)-3-hydroxy-2-methyl propanoate (99% ee) was purchased from Sigma-Aldrich and used without further purification.
18.
The reaction mixture was analyzed by thin layer chromatography (TLC, 250 μm silica gel on glass, hexanes/ethyl acetate = 9:1); visualization was accomplished with 254 nm UV light and anisaldehyde stain with heating: R
f (starting alcohol) = 0.08; R
f (benzyl ether product) = 0.36; R
f (dibenzyl ether by-product) = 0.61.
19.
The checkers utilized 120 g of silica gel (60Å, 230–400 mesh) loading the sample neat and used a gradient (19:1 heptanes:EtOAc to elute Bn
2O, to 9:1 heptanes:EtOAc to elute the desired product). The submitters utilized a silica gel column (16 cm length × 4.5 cm width, 90 g of Silica Gel 60 Geduran® 40–63 μM) wet-loaded as a slurry, loading the sample in a minimum volume of eluent, and eluting the product with 750 mL 19:1 hexanes:EtOAc.
20.
Careful analysis of the
1H NMR spectrum prior to chromatography and LC/MS analysis of the purified product indicated that impurities were formed from bis- and tris-benzylation (i.e. M+Bn and M+2Bn), presumably by addition to the aromatic ring of the primary benzyl ether product. These impurities seemed to have a greater response factor than the desired product, as their apparent level by LC/MS analysis was significantly higher than that indicated by
1H NMR or elemental analysis. For example, the elemental analysis in
Note 21 corresponded to less than 5% of the bis-benzyl product (C
19H
22O
3, vs. C
12H
16O
3 for the desired), and the
1H NMR spectrum showed no discernable quantities of any impurity. This level of impurity should not be an issue for the vast majority of applications of this methodology, but the readers should be aware of the potential presence of these side products.
21.
The compound had the following physical data, which matched the data in the literature.
4 1H NMR
pdf (400 MHz, CDCl
3) δ:1.16 (d,
J = 7.1 Hz, 3 H), 2.79 (X of ABXY
3, ddq,
JBX = 5.9 Hz,
JAX = 7.4 Hz,
JXY = 7.1 Hz, 1 H), 3.47 (B of ABXY
3, dd,
JAB = 9.1 Hz,
JBX = 5.9 Hz, 1 H), 3.64 (A of ABXY
3, dd,
JAB = 9.1 Hz,
JAX = 7.4 Hz, 1 H), 3.68 (s, 2H), 4.50 (B of AB,
J = 12.4 Hz, 1 H), 4.51 (A of AB,
J = 12.4 Hz, 1 H), 7.26–7.35 (m, 5 H);
13C NMR
pdf (100 MHz, CDCl
3) δ:13.9, 40.1, 51.7, 71.9, 73.0, 127.51, 127.53, 128.3, 138.1, 175.2; IR (thin film, cm
−1): 1736, 1454, 1363, 1198, 1176, 1092, 1028, 736, 698, 608. Anal: Calc'd for C
12H
16O
3: C, 69.21; H, 7.74. Found: C, 69.57; H, 7.86. [α]
D −10.4° (c 2.8, CHCl
3); lit [α]
D −10.6° (c 1.7, CHCl
3).
5 R
f = 0.36 (250 μm silica gel on glass, hexanes/ethyl acetate = 9:1). Chiral HLPC analysis (Chiracal OD-H 0.46 × 25 cm, 98:2 heptane:2-propanol mobile phase at 1.5 mL/min flow) indicated >99% ee. The (
R) enantiomer eluted at 3.88 min; the (
S) enantiomer eluted at 4.17 min.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Benzyl ethers are important protecting groups in organic synthesis due to their stability and the minimal electronic impact that they impart on the oxygen atom to which they are attached.
6 For example, benzyl ethers do not interfere with chelation-controlled addition of external nucleophiles to chiral aldehydes, in contrast to silyl ethers.
7 Benzyl (and modified arylmethyl) ethers are perhaps the most versatile of the alkyl ethers with respect to modes of cleavage, which include hydrogenolysis, oxidation, dissolving metal reduction, and acidic decomposition under a range of experimental protocols.
Standard conditions for the formation of benzyl ethers from the corresponding alcohols fall into two main categories: (1) the Williamson ether synthesis, an S
N2-type reaction between benzyl bromide and alkali metal alkoxides, and (2) coupling using benzyl trichloroacetimidate, which is generally promoted by trifluoromethanesulfonic acid (triflic acid, TfOH).
8 Typical benzylation reactions are thus limited to substrates that tolerate either strongly acidic or basic conditions.
2-Benzyloxy-1-methyl-pyridinium trifluoromethanesulfonate (
1) provides benzyl ethers upon warming in the presence of a free alcohol.
9 The overall balanced equation for the benzylation of alcohols (
2 →
3) is shown in Eq 1. In contrast to activated acetimidates, oxypyridinium salt
1 is stable to storage and handling. At room temperature,
1 is soluble in chlorinated solvents (dichloromethane, chloroform, dichloroethane), partially soluble in ethereal solvents (THF and ether), and insoluble in aromatic hydrocarbons (benzene, toluene). However, mixtures of
1 and aromatic solvents become homogeneous at elevated temperatures.
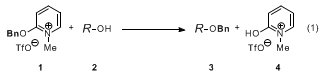
Table 1 illustrates the benzylation reactions of representative alcohols. Primary (entries 1–4) and secondary (entries 5–6) alcohols and certain tertiary alcohols (entry 7) provided the desired benzyl ethers (
3a-g) in good to excellent yield. Note that phenols are not included in Table 1; aromatic alcohols have yet to be demonstrated as viable substrates in this reaction. However, Mitsunobu conditions
10 can be applied to the benzylation of most phenols.
In conclusion, 2-benzyloxy-1-methylpyridinium triflate (1) is a novel benzylation reagent for alcohols. Salt 1 is easy to prepare, bench-stable, and pre-activated. No acidic or basic promoters are needed for benzyl transfer, which occurs upon warming in the presence of the alcohol substrate under mild conditions. New arylmethylation reagents should find use in organic synthesis for the protection of sensitive alcohol substrates.
Table 1. Scope of the Benzylation Reaction
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
Benzyl alcohol:
Benzenemethanol; (100-51-6)
2-Chloropyridine; (109-09-1)
2-Benzyloxypyridine:
Pyridine, 2-(phenylmethoxy)-; (40864-08-2)
2-Benzyloxy-1-methylpyridinium trifluoromethanesulfonate; (882980-43-0)
Methyl trifluoromethanesulfonate:
Methyl trifluoromethanesulfonate; (333-27-7)
Methyl (R)-(-)-3-benzyloxy-2-methyl propanoate:
Propanoic acid, 2-methyl-3-(phenylmethoxy)-, methyl ester, (2R)-; (112068-34-5)
Magnesium oxide; (1309-48-4)
Methyl (R)-(-)-3-hydroxy-2-methyl propionate; Propanoic acid, 3-hydroxy-2-methyl-, methyl ester, (2R)-; (72657-23-9)
 |
Gregory B. Dudley was born in 1974 in Chicago and raised in Miami, FL. Despite a vow never to take chemistry again after high school, GBD got a PhD in the subject at MIT from Rick Danheiser and was an NIH Postdoctoral Fellow with Sam Danishefsky. In 2002, Dudley returned to his alma mater, FSU, as an assistant professor. Professor Dudley (who still goes by “Greg” amongst his closest family) enjoys Costa Rican coffee, armchair philosophy, and synthetic organic chemistry.
|
 |
Kevin Wing C. Poon received his B.S. degree in 1996 and M.S. degree in 1998. He then continued his graduate studies with Professor Paul R. Hanson at the University of Kansas and received his Ph.D. degree in Chemistry in 2004. After graduation he moved to the Florida State University for his postdoctoral studies with Professor Gregory B. Dudley. Currently, he is a postdoctoral researcher in the medicinal chemistry department at the University of Kansas where his research focuses on combinatorial chemistry.
|
 |
Philip A. Albiniak was born in Myrtle Beach, SC and received B.S. degrees in chemistry and biochemistry from the College of Charleston in 2000. Later that fall, he moved to Princeton University to begin graduate studies in chemistry under the direction of Martin F. Semmelhack. In the fall of 2006 he moved to Florida State University to begin a postdoctoral association with Greg Dudley. His current research focuses on the synthetic applications of oxypyridinium salts as well as the total synthesis of roseophilin.
|
 |
Marty Berliner was born in Denver, Colorado in 1968. He received a B.S. in Physics from Harvey Mudd College in 1990 and then switched fields to organic chemistry in graduate school, where he earned a Ph.D. with Tarek Sammakia (University of Colorado, Boulder) developing the asymmetric Diels-Alder reaction of oxocarbenium ions (1996). Following postdoctoral research with David Williams at Indiana University, he joined Pfizer in 1999 and is currently a senior principal scientist in the early candidate group in Chemical Research and Development.
|
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved