Org. Synth. 2008, 85, 138
DOI: 10.15227/orgsyn.085.0138
PREPARATION OF CYCLOHEPTANE-1,3-DIONE VIA REDUCTIVE RING EXPANSION OF 1-TRIMETHYLSILYLOXY-7,7-DICHLOROBICYCLO[3.2.0]HEPTAN-6-ONE
[Cycloheptane-1,3-dione]
Submitted by Nga Do, Ruth E. McDermott, and John A. Ragan
1a.
Checked by Andrew Martins and Mark Lautens.
1. Procedure
A. 1-Trimethylsilyloxy-7,7-dichlorobicyclo[3.2.0]heptan-6-one (Note 1). A 500-mL, three-necked, round-bottomed flask equipped with a nitrogen outlet, internal temperature probe, 125-mL addition funnel (capped with a septum and nitrogen inlet), and a 65 × 20 mm egg-shaped magnetic stir bar is purged with nitrogen, then charged with 1-trimethylsilyloxycyclopentene (20.8 g, 133 mmol) (Note 2), hexanes (208 mL), and triethylamine (22.3 mL, 16.2 g, 160 mmol, 1.2 equiv) (Note 3). The addition funnel is charged with hexanes (100 mL) and dichloroacetyl chloride (12.8 mL, 19.6 g, 133 mmol, 1.0 equiv), and this solution is added dropwise to the vigorously stirred reaction mixture at a rate that maintains effective stirring and an internal temperature below 30 °C (the addition requires 30-40 min). A white precipitate (Et3N·HCl) forms during the addition, turning to a brown slurry upon complete addition. The slurry is stirred for 12 h, at which time GC analysis shows approximately 10% starting enol ether (Note 4). An additional portion of dichloroacetyl chloride (1.5 mL, 2.3 g, 16 mmol, 0.12 equiv) in 3 mL of hexanes is added to the reaction mixture via the addition funnel. After 1 h (Note 5), GC/MS analysis shows no remaining starting material. A sintered-glass Büchner funnel (8 cm diameter, 350 mL capacity) is charged with 40 g of silica gel (Note 6) slurried in hexanes, and the reaction mixture is filtered with suction through this pad into a 2-L filter flask rinsing with an additional 600 mL of hexanes in three portions. The clear, pale-yellow filtrate is concentrated on the rotary evaporator (25 mmHg, room temperature water bath) to provide the product as a pale-yellow liquid which crystallizes to a yellow solid when stored in the freezer (7 °C ) (29.9 g, 112 mmol, 84% yield) (Note 7). This material is used without further purification in the next step.
B. 1-Trimethylsilyloxybicyclo[3.2.0]heptan-6-one. A 1-L, four-necked, round-bottomed flask equipped with an overhead stirrer, adapter with internal temperature probe, reflux condenser with nitrogen inlet, and a septum with nitrogen outlet is purged with nitrogen, then charged with 2-propanol (350 mL, Note 3) and nitrogen is bubbled through the solvent for 15 min via a 30-cm, 18 gauge needle. The mechanical stirrer, fitted with a 60 X 20 mm Teflon paddle, is started at 120–150 rpm. 1-Trimethylsilyloxy-7,7-dichlorobicyclo[3.2.0]heptan-6-one (29.9 g, 112 mmol) is dissolved in 50 mL of 2-propanol (gentle heating is required) and added to the reaction flask, rinsing with an additional 50 mL, then 20 mL of 2-propanol. Nitrogen is then bubbled through the solution for 15 min. The septum is removed and replaced with a glass funnel, through which 10% Pd/C (5.98 g, 50% w/w in water) is added, the septum is replaced, and nitrogen is bubbled through the solution for 5 min. The septum is removed again, replaced with a glass funnel, and sodium formate is added in a single portion (38.1 g, 559 mmol, 5.0 equiv, Note 3), rinsing with 25 mL of 2-propanol The septum is replaced and nitrogen is bubbled through the solution for 5 min. The flask is heated in an oil bath to an internal temperature of 80 °C, and is maintained at this temperature with stirring for 18 h. The reaction mixture is cooled to room temperature and a 60-μL aliquot is removed and diluted with 600 μL of ether. A 4-μL sample of this solution is analyzed by GC (Note 4), and shows complete conversion. The slurry is filtered with suction through a pad of Celite (8 cm diameter sintered glass funnel, 350 mL volume, with a 4 cm pad of Celite), into a 1-L filter flask rinsing with four 50-mL portions of 2-propanol. The filtrate is concentrated on the rotary evaporator (25 mmHg, room temperature water bath) to a cloudy, yellow oil, which is then diluted with 150 mL of methyl t-butyl ether (MTBE, Note 3) and then is transferred to a 250-mL separatory funnel. The organic phase is washed with three portions of half-saturated brine (75 mL each), once with saturated brine, then is dried over sodium sulfate, filtered, and concentrated on the rotary evaporator (25 mmHg, room temperature water bath) to provide the product as a clear, orange oil (17.2 g, 86.7 mmol, 77% yield) (Note 8) with traces of MTBE and 2-propanol. This material is used without further purification in the next reaction.
C. Cycloheptane-1,3-dione. A three-necked, 250-mL, round-bottomed flask equipped with an internal temperature probe, 50-mL addition funnel with nitrogen inlet, a septum with nitrogen outlet and magnetic stir bar is purged with nitrogen and charged with 1-trimethylsilyloxybicyclo[3.2.0]-heptan-6-one (17.1 g, 81.0 mmol) and 33 mL each of 2-propanol and water. The solution is cooled to an internal temperature of 0-5 °C with an ice-water bath, and 26 mL of 2:1 water-acetic acid (v/v) is added dropwise via the addition funnel over 30 min. At this rate of addition, the internal temperature remains below 5 °C throughout the addition. The solution is then allowed to stir for 16 h and gradually come to room temperature. A 60-μL aliquot is removed and diluted with 600 μL of ether from which a 4-μL sample is used for GC analysis (Note 4), which shows complete conversion to cycloheptane-1,3-dione. The reaction mixture is poured into 200 mL of MTBE in a 500-mL separatory funnel. The layers are separated, and the aqueous phase is extracted with an additional 100 mL of MTBE. The organic solutions are combined, washed with 50 mL of brine, and dried over sodium sulfate. Filtration through cotton, rinsing with 50 mL MTBE and concentration on the rotary evaporator (25 mmHg, 30 °C water bath) provides the crude product as a clear, amber oil. 1H NMR analysis shows the diketone contaminated with residual MTBE and acetic acid. The oil is dissolved in 50 mL of dichloromethane, and treated with 22.5 g of silica gel (Note 6). Concentration on the rotary evaporator provides a tan, free-flowing solid, which is placed on the top of a 7.5 × 23 cm column of silica gel packed as a slurry in hexane/Et2O, 2:1. The column is eluted with hexane/Et2O, 2:1 (500 mL) and 1:1 (3.8 L), followed by combination of the product-containing fractions (TLC in hexane/Et2O, 1:1, visualization with p-anisaldehyde stain). The combined fractions are concentrated via rotary evaporator, under vacuum (25 mmHg, room temperature water bath) to provide cycloheptane-1,3-dione as a clear, pale-yellow oil (6.69 g, 53.1 mmol, 61% yield). This material is approximately 95% pure as determined by 1H NMR analysis and is suitable for most subsequent uses, although the chromatographed material fails combustion analysis. If necessary, further purification can be achieved by short path vacuum distillation (Notes 9, 10, and 11).
2. Notes
1.
This procedure is a modification of that reported previously by the submitters.
32.
1-Trimethylsilyloxycyclopentene (97%) was purchased from Aldrich Chemical Company, Inc., and used as received. It can also be prepared from cyclopentanone.
4 The reported yields were obtained using a freshly-opened bottle; older bottles became colored and cloudy, and afforded lower yields.
3.
Dichloroacetyl chloride (98%, freshly opened bottle), methyl t-butyl ether (99.8%, HPLC grade) and sodium formate (99+%) were purchased from Aldrich Chemical Company.
Glacial acetic acid (99%), hexane (99%) and isopropyl alcohol (99.5%) were purchased from Fisher Scientific.
Triethylamine (98%) was purchased from ACP Chemicals.
Pd/C (10 wt%) was purchased from Alfa Aesar.
All reagents and solvents were used as received.
4.
A Hewlett-Packard GC with an HP-5 0.25 mm × 60 m, 0.25 μm column was used. Oven temperature program: 1 min at 40 °C, 20 °C/min ramp to 300 °C, 4 min hold at 300 °C. The carrier gas (He) was held at a constant flow rate of 1.0 mL/min. Detection was by flame ionization detector.
|
| Compound |
Retention time (min) |
|
|
1-Trimethylsilyloxycyclopentene
|
9.1
|
|
1-Trimethylsilyloxy-7,7-dichlorobicyclo[3.2.0]heptan-6-one (1)
|
12.9
|
|
1-Trimethylsilyloxy-bicyclo[3.2.0]heptan-6-one (2)
|
11.4
|
|
Cycloheptane-1,3-dione (3)
|
10.9
|
|
5.
GC analysis after 30 minutes still showed a peak for the silyl enol ether (~7% remaining); however, the addition of extra Et
3N and/or dichloroacetyl chloride did not change the product/silyl enol ether ratio.
6.
Silica gel (40-63 μm) was purchased from Silicycle Inc.
7.
1H NMR
pdf (CDCl
3, 400 MHz) δ: 0.25 (s, 9 H), 1.50-1.63 (m, 1 H), 1.86-1.98 (m, 2 H), 2.00-2.11 (m, 2 H), 2.51-2.58 (m, 1 H), 3.66 (br d,
J = 8.6 Hz, 1 H);
13C NMR
pdf (100 MHz, CDCl
3) δ 1.6, 25.2, 29.1, 38.1, 67.9, 87.8, 92.2, 199.3; IR thin film (cm
−1) 844, 1106, 1253, 1324, 1804, 2961; MS (EI)
m/z (relative intensity): 266 (1), 149 (20), 93 (12), 84 (12), 79 (17), 75 (12), 73 (100), 55 (12). This data is in agreement with that reported by Krepski and Hassner.
58.
1H NMR
pdf (CDCl
3, 400 MHz) δ: 0.17 (s, 9 H), 1.47-1.65 (m, 1 H), 1.76-2.00 (m, 4 H), 2.11-2.18 (m, 1 H), 2.92-3.01 (m, 1 H), 3.24-3.39 (m, 2 H);
13C NMR
pdf (100 MHz, CDCl
3) δ 1.5, 25.8, 28.7, 40.5, 59.7, 70.3, 77.4, 212.4; IR neat (cm
−1) 844, 1087, 1224, 1253, 1782, 2956MS (EI)
m/z (relative intensity): 170 (18), 169 (100), 157 (75), 156 (95), 155 (25), 75 (90), 73 (68). This data is in agreement with that reported by Pak.
69.
The chromatographically-purified
diketone (3.00 g) is placed in a 25-mL round bottomed flask equipped with a short-path distillation apparatus (8-cm stillhead length with a 7-cm jacketed water-cooled condenser and 4 pronged distribution adapter) and heated in an oil bath (bath temperature reached ~105 °C) under reduced pressure (0.20 - 0.22 mmHg). The product distilled over at a head temperature of 81-82 °C, to afford 2.34 g (78 % recovery) of analytically pure diketone
(Note 10).
10.
The product exhibits the following physicochemical properties:
1H NMR
pdf (CDCl
3, 400 MHz) δ: 1.96-2.03 (m, 4 H), 2.56-2.63 (m, 4 H), 3.61 (s, 2 H);
13C NMR
pdf (CDCl
3, 100 MHz) δ: 25.1, 44.2, 59.9, 205.1; MS (EI)
m/z (relative intensity): 126 (68), 98 (100), 83 (37), 70 (43), 55 (49); IR (film): 3608 , 2943, 2867, 1714, 1694, 1455, 1207, 1134, 924 cm
−1. HRMS (EI): calcd for C
7H
10O
2: 126.0681 (M
+), found: 126.0679. Anal. Calcd for C
7H
10O
2: C, 66.65; H, 7.99; Found: C, 66.34; H, 8.14. This data is in agreement with that reported previously.
2f11.
The diketone should be stored cold under nitrogen. The diketone is unstable to base, readily undergoing a retro-Dieckmann cyclization to form 6-oxoheptanoic acid.
7 The checkers report that storing under nitrogen is not necessary, and that the product did not noticeably degrade after several months in the freezer at -10 °C.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Several syntheses of cycloheptane-1,3-dione have been reported in the literature.
2 At the time this work was initiated, we required a practical synthesis capable of providing multi-kilogram quantities of this intermediate. None of the existing literature syntheses were deemed adequate for this purpose due to a combination of practicality considerations and the use of heavy metal or potentially explosive reagents (e.g. ethyl diazoacetate,
2a PhHg(CBr
3),
2c ClCH
2OCH
3,
2e or Hg(OAc)
2).
2f We were aware of Hassner's preparation of the bicyclic adduct of dichloroketene and 1-trimethylsilyloxycyclopentene,
5 and Pak's observation that the dechlorinated derivative of this diketone was converted to cycloheptane-1,3-dione upon treatment with fluoride ion.
6 However, the conditions described for the reduction (stoichiometric amount of Bu
3SnH) were not practical for the large scale preparation of an intermediate destined to be converted into clinical supplies. We found and reported that a Zn/AcOH/aq 2-propanol system effected the desired reduction, desilylation and ring expansion in a single-pot sequence.
3 Unfortunately, upon scale-up this reaction suffered a dramatic decrease in yield, due to formation of a mixture of the desired diketone and 2-acetylcyclopentanone,
7 an observation also made by others.
8We believe the reason for this formation of consitutuional isomers to be competition between two reaction pathways, as shown below.
7 In path A, reduction of both chlorine atoms prior to desilylation leads to clean formation of the desired diketone (
3). However, if desilylation precedes chlorine atom reduction (path B), then the retro-aldol reaction generates a 2-acetyl-substituted cyclopentanone (
4) by rupture of the C
1-C
7 bond. Chlorine atom reduction then generates 2-acetylcyclopentanone (
5). These results are consistent with observations made by Hassner and Krepski on related dichlorocyclobutanones.
5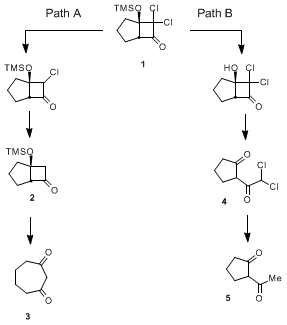
In this improved procedure we report that transfer hydrogenolysis with sodium formate efficiently reduces the chlorine atoms without any desilylation. Acid-mediated desilylation then cleanly forms the desired diketone, with none of the isomeric product being formed. Although this improved process involves three separate operations, there is no need to purify either intermediate, leading to an efficient preparation of the title diketone.
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
Cycloheptane-1,3-dione (1194-18-9)
1-Trimethylsilyloxybicyclo[3.2.0]heptan-6-one (125302-44-5)
1-Trimethylsilyloxy-7,7-dichlorobicyclo[3.2.0]heptan-6-one (66324-01-4)
Dichloroacetyl chloride (79-36-7)
1-Trimethylsilyloxycyclopentene (19980-43-9)
Acetic acid (64-19-7)
 |
John Ragan was born and raised in Kansas City, Missouri. He received his B.S. degree from MIT in 1985, where he performed undergraduate research with Rick Danheiser supporting the total synthesis of anatoxin a. He then joined Stuart Schreiber's group at Yale University, moving with Stuart to Harvard in 1988. He completed his Ph.D. in 1990 on the total synthesis of FK506. He then joined Clayton Heathcock's group at the University of California-Berkeley as an American Cancer Society podstodoctoral fellow, where he worked on the biomimetic synthesis of daphniphyllum alkaloids. He joined Pfizer Global Research & Development in 1992, where he has worked in Discovery Chemistry and Chemical Research & Development. He is currently an Associate Research Fellow. Outside of work he enjoys jazz trumpet and Texas Hold 'Em poker. |
 |
Nga Do was born in 1973 and grew up in Indiana. She attended Purdue University where she earned a B.S. in Chemistry and conducted undergraduate research in the labs of Professor Mark Lipton. She then joined Professor Scott Rychnovsky's lab at the University of California, Irvine, receiving her M.S. in 1998.
She then joined the Chemical Research and Development group at Pfizer in Groton, Connecticut. |
 |
Ruth McDermott was born in Boston, MA, and received a B.S. in Chemistry from the University of Massachusetts at Boston. She completed her undergraduate research under the direction of J.-P. Anselme. She is currently part of the Chemical Research and Development group at Pfizer in Groton, CT, where she has been employed for 20 years. She enjoys photography and participating in short-term mission trips in the US and abroad. |
 |
Andrew Martins was born in Toronto, Ontario, Canada, and raised in nearby Brampton, Ontario. His post-secondary education began at the University of Waterloo where he completed the Honours Co-operative Chemistry program and performed his honours B.Sc. thesis work in the laboratories of Professor Éric Fillion. He then joined the group of Professor Mark Lautens at the University of Toronto where he is currently a graduate student. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved