Org. Synth. 2008, 85, 147
DOI: 10.15227/orgsyn.085.0147
SYNTHESIS OF ENANTIOPURE DI(TERT-BUTYL) (2S,4S)-4-HYDROXY-6-OXO-1,2-PIPERIDINEDICARBOXYLATE. A USEFUL BUILDING BLOCK FOR THE PREPARATION OF 4-HYDROXYPIPECOLATE DERIVATIVES
Submitted by Olivier Chaloin
1, Frédéric Cabart
2, Julien Marin
1,3, Haixiang Zhang
2, and Gilles Guichard
1.
Checked by Masakatsu Shibasaki and Hisashi Mihara.
1. Procedure
A. 2-tert-Butoxycarbonylamino-4-(2,2-dimethyl-4,6-dioxo-[1,3]dioxan-5-yl)-4-oxo-butyric acid tert-butyl ester. A one-necked, 500-mL, round-bottomed flask containing a magnetic stir bar and fitted with a three-way stopcock fitted with a rubber septum and an argon inlet is charged with 15.0 g (51.85 mmol) of N-α-tert-butoxycarbonyl-L-aspartic acid α-tert-butyl ester (Boc-L-Asp-Ot-Bu) (Note 1) and 150 mL of dichloromethane (Note 2). The resulting solution is cooled in an ice bath at 0 °C (ice bath temperature) whereupon 7.85 g (54.44 mmol, 1.05 equiv) of 2,2-dimethyl-1,3-dioxane-4,6-dione (Meldrum's acid) (Note 3) and 9.50 g (77.78 mmol, 1.5 equiv) of 4-dimethylaminopyridine (DMAP) (Note 4) are added. N-(3-Dimethylaminopropyl)-N'-ethylcarbodiimide hydrochoride (EDC·HCl) (14.91 g, 77.78 mmol, 1.5 equiv) (Note 5) is then added portion-wise to the reaction mixture at 0 °C over 5 min. The three-way adapter is returned to the neck and reaction mixture is allowed to warm to room temperature and is stirred under argon for 3 h (Note 6). The solution is diluted with 50 mL of dichloromethane before being transferred to a 500-mL separatory funnel and is then washed successively with two, 150-mL portions of 1 M aq KHSO4 solution and 150 mL of brine. The organic layer is dried over Na2SO4 (25–30 g), then is filtered through a cotton plug in a funnel, collected in a 500-mL flask and concentrated by rotary evaporation (40 °C, 6 mm Hg) to provide 21.5 g of crude 2 as a clear, pale-yellow oil (Note 7).
B. Di(tert-butyl) (2S)-4,6-dioxo-1,2-piperidinedicarboxylate. Ethyl acetate (330 mL) (Note 8) is added to the 500-mL flask containing 2, which is then equipped with a magnetic stir bar and a reflux condenser. The solution is heated in an oil bath at 80 °C (bath temperature) for 3 h (Note 9). The solution is allowed to cool before being transferred to a 1-L separatory funnel and is washed successively with 200 mL of 1 N aq KHSO4 solution and 200 mL of brine. The organic layer is dried over Na2SO4 (25–30 g), filtered through a cotton plug in a funnel, and concentrated by rotary evaporation (34–40 °C, 6 mm Hg). The residual crystalline material is slurried in 100 mL of cyclohexane, then is collected by filtration in a Hirsch funnel and is washed with two, 50-mL portions of cyclohexane and then dried under reduced pressure (40 °C, 6 mm Hg) to afford 12.61 g (78%, 2 steps from 1) of dioxopiperidine 3 as a white solid (Notes 10, 11).
C. Di(tert-butyl) (2S,4S)-4-hydroxy-6-oxo-1,2-piperidinedicarbox-ylate. A two-necked, 500-mL, round-bottomed flask fitted with a rubber septum, argon needle inlet and thermometer is charged with dioxopiperidine 3 (12.61 g, 40.25 mmol) and 150 mL of dichloromethane (Note 12). The resulting solution is cooled in an ice bath at 0 °C (bath temperature) while 25 mL of AcOH (Note 13) is added. Sodium borohydride (NaBH4) (4.57 g, 120.75 mmol, 3.0 equiv) (Note 14) is then added portion-wise to the reaction mixture at such a rate as to keep the internal temperature below 10 °C. The reaction mixture is allowed to warm to room temperature and is stirred for 12 h (Note 15). The reaction mixture is neutralized with 150 mL of 1 M aq KHSO4 solution and then is transferred to a 500-mL separatory funnel. The aqueous phase is separated and the organic layer is dried over Na2SO4 (25–30 g), filtered through a cotton plug in a funnel, and concentrated by rotary evaporation (40 °C, 6 mm Hg). Dichloromethane (250 mL) (Note 12) is added to the 500-mL flask, which is then equipped with a magnetic stir bar. The resulting solution is cooled in an ice bath at 0 °C while 25 mL AcOH (Note 13) is added. Sodium borohydride (NaBH4) (1.52 g, 40.25 mmol, 1.0 equiv) (Note 14) is then added portion-wise to the reaction mixture over 5 min. The reaction mixture is allowed to warm to room temperature and is stirred for 4 h. The reaction mixture is neutralized with 100 mL of a 1 M aq KHSO4 solution and is transferred to a 500-mL separatory funnel. The aqueous phase is separated and the organic layer is dried over Na2SO4 (25–30 g), filtered through a cotton plug in a funnel, and concentrated by rotary evaporation (40 °C, 6 mm Hg) (Notes 16, 17). The residue is dissolved in 200 mL of ethyl acetate, and the solution transferred to a 500-mL separatory funnel where it is washed successively with two, 150-mL portions of 1 M aq KHSO4 solution and two, 150-mL portions of sat. aq sodium bicarbonate solution and 150 mL of brine. The organic layer is dried over Na2SO4 (25–30 g), filtered through a cotton plug in a funnel, and concentrated by rotary evaporation (40 °C, 6 mm Hg). The residual crystalline solid is dissolved in 23 mL of dichloromethane. Diisopropyl ether (150 mL) is slowly added and the resulting slurry is triturated for 1 h. The white solid that precipitates is collected by filtration, then is washed with 50 mL of diisopropyl ether, 50 mL of cyclohexane and then is dried under reduced pressure (25 °C, 6 mm Hg and then 0.04 mm Hg) to afford 6.25 g (49.3%) of 4. The filtrate is concentrated and the residue is dissolved in 9 mL dichloromethane. After addition of diisopropyl ether (100 mL) and a magnetic stir bar, the mixture is stirred for 10 h at room temperature to afford a second crop (1.60 g), which is collected by filtration and is combined with the first crop of crystals to give a final yield of 7.85 g (62 %) (Note 18).
2. Notes
1.
N-α-tert-butoxycarbonyl-L-aspartic acid α-tert-butyl ester (98+%), was purchased from Novabiochem by the submitters and from Waterstone Technology Product List by the checkers, was used as received.
2.
Dichloromethane was purchased from WAKO Pure Chemicals and was used as received.
3.
2,2,5-Trimethyl-1,3-dioxane-4,6-dione (Meldrum's acid) (98%) was purchased from Aldrich Chemical Co., Inc and was used as received.
4.
4-DMAP (99%) was purchased from Aldrich Chemical Co., Inc and was used as received.
5.
N-(3-Dimethylaminopropyl)-N'-ethylcarbodiimide hydrochoride (EDC·HCl) was purchased from Senn Chemicals by the submitters and from Watanabe Chemical Industries, LTD by the checkers, was used as received.
6.
The progress of the reaction was followed by TLC analysis on silica gel with cyclohexane/ ethyl acetate/acetic acid, 7:3:0.5) as eluent. Visualization of the TLC plates was performed with ninhydrin. Boc-Asp-O
t-Bu has R
f = 0.31 (red) and product
2 has R
f = 0.20 (dark red).
7.
Compound
2 is used in the next step without further purification. The submitters observed partial spontaneous rearrangement to dioxopiperidine
3 upon drying compound
2 under reduced pressure for 5 days. Compound
2 has the following spectroscopic properties:
1H NMR
pdf (500 MHz, CDCl
3,) δ: 1.42 (s, 9 H), 1.44 (s, 9 H), 1.74 (s, 3 H), 3.51 (m, 1 H), 3.66 (m, 1 H), 4.67 (m, 1 H), 5.27 (br, 1 H).
8.
Ethyl acetate (puriss. p.a. ACS reagent grade) was purchased from Aldrich Chemical Co., Inc and used as received.
9.
The progress of the reaction was followed by TLC analysis on silica gel with cyclohexane/ethyl acetate/acetic acid, 7:3:0.5 as eluent. Visualization of the TLC plates was performed with ninhydrin; dioxopiperidine
3 has R
f = 0.29 (light orange).
10.
Dioxopiperidine
3 exhibits the following physicochemical properties: C
18 RP-HPLC
tR 23.4 (HPLC analysis was performed on a CAPCELL PAC C
18 column (5 μm, 4.6 mm × 250 mm) (
SHISEIDO) by using a linear gradient of A (0.1% TFA in H
2O) and B (0.08% TFA in CH
3CN) at a flow rate of 1.2 mL/min with UV detection at 214 nm; linear gradient, 20-80% B, 20 min); colorless crystals; [α]
D23 = +107.2 (c = 1.02, CHCl
3); mp 114-116 °C;
1H NMR
pdf (500 MHz, CDCl
3) δ: 1.46 (s, 9 H), 1.55 (s, 9 H), 2.83 (dd,
J = 17.7, 7.0 Hz, 1 H), 3.03 (dd,
J = 17.7, 2.1 Hz, 1 H), 3.37 (d,
J = 19.5 Hz, 1 H), 3.53 (d,
J = 19.6 Hz, 1 H), 5.07 (dd,
J = 7.1, 2.2 Hz, 1 H);
13C NMR
pdf (125 MHz, CDCl
3) δ: 27.8 (3 CH
3), 27.9 (3 CH
3), 40.9 (CH
2), 50.4 (CH
2), 54.6 (CH), 84.0 (C), 84.7 (C), 151.2 (C), 165.3 (C), 168.9 (C), 200.2 (C); IR (KBr) 2981, 1766, 1725, 1644, 1600, 1278, 1147 cm
−1; ESI-MS [M
++Na] 336. Anal. Calcd for C
15H
23NO
6: C, 57.50; H, 7.40; N, 4.47. Found: C, 57.22; H, 7.32; N, 4.38.
11.
Dioxopiperidine
3 exists in equilibrium with di(
tert-butyl) (2
S)-4-hydroxy-6-oxo-3,6-dihydro-1,2(2
H)-pyridinedicarboxylate, the thermo-dynamically stable enol form. The product
3 exists exclusively in the keto form in CDCl
3, whereas in DMSO-
d6, the
1H and
13C NMR spectra show the enol form only:
1H NMR
pdf (500 MHz, (CD
3)
2SO)
δ: 11.2 (s, 1H), 4.95 (d,
J = 1.6 Hz, 1H), 4.85 (dd,
J = 6.9, 1.5 Hz, 1H), 3.04 (ddd,
J = 17.7, 6.9, 1.6 Hz, 1H), 2.59 (dd,
J = 17.7, 1.5 Hz, 1H), 1.46 (s, 9H), 1.41 (s, 9H);
13C NMR
pdf (125 MHz, (CD
3)
2SO)
δ: 169.6, 168.2, 164.1, 152.0, 96.2, 81.6, 81.4, 55.0, 30.3, 27.7, 27.5.
12.
Dichloromethane was freshly distilled over from calcium hydride under argon.
13.
AcOH (99.5+%), purchased from Prolabo by the submitters and from Wako Pure Chemical Industries, Ltd. by the checkers), was used as received.
14.
Sodium borohydride (NaBH4) (98+%) was purchased from Acros Organics by the submitters (KANTO Chemical CO., Inc.) and was used as received.
15.
At that stage, C
18 RP-HPLC analysis of the reaction mixture reveals that 11% of dioxopiperidine
3 is still present (see
Note 16 for analysis conditions). On this scale, as well as on larger scale, the submitters found that a prolonged reaction time was not sufficient to bring the reaction to completion and that satisfactory results were obtained by performing the reduction a second time after work-up.
16.
Analytical C
18 RP-HPLC: CAPCELL PAC C
18 column (5 μm, 4.6 mm × 250 mm) (
SHISEIDO) (linear gradient, 20-80% B, 20 min) of the crude product reveals that the reduction is complete (< 2 % of
3) and that the diastereomeric ratio between di(
tert-butyl) (2
S,4
S)-4-hydroxy-6-oxo-1,2-piperidinedicarboxylate (
4) and di(
tert-butyl) (2
S,4
R)-4-hydroxy-6-oxo-1,2-piperidinedicarboxylate (
epi-4) is 91:9. The elution times (
tR) for
3,
4 and
epi-4 are 23.4, 20.6, and 21.4 min, respectively.
17.
After this workup, the product still contained acetic acid which was detrimental to the recrystallization. A second extractive workup as described was needed to remove all of the acetic acid.
18.
(2
S,4
S)-4-Hydroxy-6-oxo-1,2-piperidinedicarboxylate
4 was obtained in high diastereomeric purity (dr > 99:1 for the combined crops as determined by analytical C
18 RP-HPLC (linear gradient, 20-80% B, 20 min))
(Note 10). The enantiomeric ratio was > 99:1 as determined by HPLC analysis on an OJ-H column (5 μm, 3.9 m × 150 mm) by elution with 5% isopropanol in
n-hexane at a flow rate of 1 mL/min with UV detection at 214 nm. The elution times (
tR) for
4 and
ent-4 are 11.8 and 8.4 min, respectively. Lactam
4 exhibited the following physicochemical properties: C
18 RP-HPLC
tR 20.6 min (linear gradient, 20-80% B, 20 min); [α]
D21 = -23.7 (c = 1.02, CHCl
3); colorless crystals; mp 128-129 °C;
1H NMR
pdf (500 MHz, CDCl
3) δ: 1.46 (s, 9 H), 1.51 (s, 9 H), 2.11 (d,
J = 4.0 Hz, 1 H), 2.22 (ddd,
J = 14.1, 6.4, 2.8 Hz, 1 H), 2.38-2.43 (m, 1 H), 2.60 (ddd,
J = 17.4, 4.3, 1.9 Hz, 1 H), 2.77 (dd,
J = 17.4, 4.9 Hz, 1 H), 4.26 (m, 1 H), 4.65 (dd,
J = 6.8, 3.7 Hz, 1 H);
13C NMR
pdf (125 MHz, CDCl
3) δ: 27.8 (3 CH
3), 27.9 (3 CH
3), 32.9 (CH
2), 43.2 (CH
2), 56.1 (CH), 64.2 (CH), 82.5 (C), 83.5 (C), 151.8 (C), 168.6 (C), 171.2 (C); IR(KBr) 3504, 2975, 1766, 1706, 1284, 1139; ESI-MS [M
++Na] 338; Anal. Calcd for C
15H
25NO
6: C, 57.13; H, 7.99; N, 4.44. Found: C, 56.88; H, 7.84; N, 4.33.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Five-membered ring 2,4-dioxo-1-pyrrolidinecarboxylates (chiral tetramic acids) obtained from Meldrum's acid and
N-protected α-amino acids have received considerable attention as precursors of statine [(3
S,4
S)-4-amino-3-hydroxy-6-methylheptanoic acid] derivatives.
4-8 Their stereocontrolled reduction leads exclusively to the
cis-4-hydroxy derivative. However, there have been relatively few reports on the synthesis and reactivity of
N-acylated 4,6-dioxopiperidines, their six-membered counterparts.
9By analogy with the synthesis of chiral tetramic acids,
4 condensation of
N-protected 3-aminopropanoic acids with Meldrum's acid in the presence of EDC·HCl and DMAP, followed by cyclization at 80 °C in
ethyl acetate, provides a convenient and short entry to enantiopure
N-acylated 4,6-dioxopiperidines (e.g.
3).
10 N-Acylated 4,6-dioxopiperidines exist in equilibrium with the thermodynamically stable enol form.
We found that treatment of various
N-acylated 4,6-dioxopiperidines with NaBH
4 in CH
2Cl
2/AcOH (10:1) for 16-72 h resulted in a quantitative and stereoselective reduction of the keto functionality.
10 The selectivity of the reaction was significantly influenced by the bulk of the side chain at C2, the lowest and highest selectivities being observed for the methyl (dr 84:16) and isopropyl groups (dr >99:1), respectively. In the case of a carboxylate side chain at C2, the
tert-butyl ester group (in
3) exerts a stronger stereodirecting effect than the corresponding benzyl ester. The stereoselective reduction in step (C) afforded
4 in diastereomerically pure form (dr >99:1) following a single recrystallizaton. The absolute configuration at C4 was confirmed by X-ray crystal structure determination.
10This three-step procedure allows for the preparation of (2
S,4
S)-4-hydroxy-6-oxo-1,2-piperidinedicarboxylate
4 in 46% overall yield from Boc-L-Asp-OtBu and using common and cheap reagents. Hydroxylated lactam
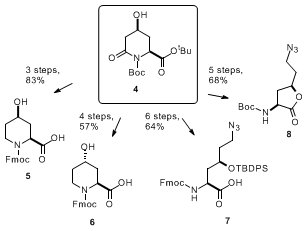
4 is a versatile intermediate that can be readily converted to several useful amino acid derivatives.
10,11 Reduction of the lactam moiety by treatment with BH
3·SMe, and subsequent deprotection/protection afforded the enantiopure protected
cis-4-hydroxypipecolic acid
5, an amino acid constituent of natural antibiotics and several drug candidates, in 83% yield. The corresponding
N-Fmoc protected
trans-(2
S,4
S)-4-hydroxypipecolate
6 was obtained by further inversion of the C4 stereocenter under Mitsunobu conditions (57% from
4). Alternatively, ring opening of
4 under reductive conditions (NaBH
4, EtOH) following protection of the hydroxyl group gave the corresponding 1,3-diol (85% (TBDPS protecting group); 83% (TBDMS protecting group)), which can be further transformed to the 4-hydroxylysine derivative
7 or to enantiomerically pure lactone
8.
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
N-α-tert-Butoxycarbonyl-L-aspartic acid α-tert-butyl ester (Boc-L-Asp-Ot-Bu); (34582-32-6)
2,2-Dimethyl-1,3-dioxane-4,6-dione (Meldrum's acid); (2033-24-1)
N-(3-Dimethylaminopropyl)-N'-ethylcarbodiimide hydrochoride (EDC·HCl); (25952-53-8)
2-tert-Butoxycarbonylamino-4-(2,2-dimethyl-4,6-dioxo-[1,3]dioxan-5-yl)-4-oxo-butyric acid tert-butyl ester; (10950-77-9)
Di(tert-butyl) (2S)-4,6-dioxo-1,2-piperidinedicarboxylate; (653589-10-7)
Sodium borohydride; (16940-66-2)
Di(tert-butyl) (2S,4S)-4-hydroxy-6-oxo-1,2-piperidinedicarboxylate; (653589-16-3)
 |
Gilles Guichard was born in Orléans (France) in 1969. He studied chemistry at the Ecole Nationale Supérieure de Chimie in Toulouse and received his Ph.D. in 1996 from the University Louis Pasteur (Strasbourg) under the guidance of Dr. Jean-Paul Briand working on the synthesis and immune recognition of pseudopeptides. After post-doctoral research with Prof. Dieter Seebach at the ETH Zürich (Switzerland) working on beta-peptides, he returned to Strasbourg as a CNRS Chargé de Recherche in the Institute of Molecular and Cellular Biology and was promoted CNRS Directeur de Recherche in 2006. His research is mainly focused on biomimetic chemistry of peptides, folding and biomolecular recognition. |
 |
Olivier Chaloin received his Ph.D. in chemistry from the University of Montpellier II in 1996. After postdoctoral studies with Prof. G. Flouret at Northwestern University (Chicago, USA) and with Prof. R. Offord in Geneva (Switzerland), he joined the CNRS and moved to the Institute of Molecular and Cellular Biology in Strasbourg (France).
His research interests focus on peptide, pseudopeptide and protein synthesis. |
 |
Frédéric Cabart was born in 1972 in France. He received his B.S. and M.S. degrees in Chemistry from the Université de Nancy I, France in 1996 and 1997 respectively. He has been employed at NeoMPS S.A. France since 1998, where he currently works as Research & Development Chemist involved in the syntheses of pharmaceutical building blocks from laboratory scale to kilogram scale. |
 |
Julien Marin was born in Metz (France) in 1976. He studied chemistry at the Ecole Nationale Supérieure de Chimie in Mulhouse and received his Ph.D. in 2003 from the University Louis Pasteur (Strasbourg) under the guidance of Dr. Gilles Guichard working on the synthesis and auto-immune recognition of glycopeptides. After post-doctoral research with Prof. Stephen Hanessian at the University of Montreal (Canada) working on the design and synthesis of new inhibitors of endothelin converting enzymes, he joined the contract research organization Novalyst Discovery in 2006. His current position is project manager in the organic custom-synthesis department. |
 |
Haixiang Zhang was born in 1963 in China. He received his Ph.D. in Organic Chemistry under the supervision of Prof. G. Balavoine and Dr. F. Guibé from the Université de Paris-Sud, France in 1990. He then joined the Propeptide/Isochem S.A. France as Research & Development Chemist. In 2000, he moved to current position as Research & Development Manager at NeoMPS S.A., France. His current interests focus on the organic synthetic chemistry in pharmaceutical industry. |
 |
Hisashi Mihara was born in 1981 in Shiga, Japan. He received his M.S. degree in 2006 from the University of Tokyo. He is pursuing a Ph.D. degree at the Graduate School of Pharmaceutical Sciences, The University of Tokyo, under the guidance of Professor Masakatsu Shibasaki. His current interest is catalytic enantioselective total synthesis of biologically active compounds. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved