Org. Synth. 2010, 87, 68
DOI: 10.15227/orgsyn.087.0068
CATALYTIC ASYMMETRIC ADDITION OF AN IN-SITU PREPARED ARYLZINC TO CYCLOHEXANECARBOXALDEHYDE: (R)-(+)-α-CYCLOHEXYL-3-METHOXY-BENZENEMETHANOL
Submitted by Albert M. DeBerardinis, Mark Turlington, and Lin Pu
1.
Checked by Kyle L. Kimmel and Jonathan A. Ellman.
1. Procedure
CAUTION! Neat diethylzinc may ignite on exposure to air and reacts violently with water. It must be handled and reacted under nitrogen. The reaction solvent must be dried and distilled prior to use and all glassware and syringes must be thoroughly dried.
An oven-dried 500-mL, 3-necked, round-bottomed flask, stir bar, glass stopper, septum and vacuum adaptor containing a stopcock are taken into a glove box (Note 1). The flask is charged with lithium acetylacetonate (0.771 g, 7.27 mmol, 0.25 equiv) (Note 2), sealed, and brought outside of the glove box and into a fume hood. The vacuum adaptor is connected to a Schlenk line, and the flask is placed under a nitrogen atmosphere (Note 3).
1-Methyl-2-pyrrolidinone (NMP, 48 mL) (Note 4) and 3-iodoanisole (7.62 mL, 64.0 mmol, 2.2 equiv) (Note 5) are added sequentially through the septum via syringe. The reaction flask is cooled to 0 ºC with an ice bath, and diethylzinc (3.65 mL, 34.9 mmol, 1.2 equiv) (Note 6) is added via syringe. The pale yellow reaction mixture is stirred at 0 ºC for 12 h.
(S)-3,3'-Bis-morpholinomethyl-5,5',6,6',7,7',8,8'-octahydro-1,1'-bi-2-naphthol [(S)-1] (Note 7) is dried prior to use in catalysis. An oven-dried 100-mL Schlenk flask equipped with a stir bar is connected to a Schlenk line, fitted with a glass stopper and cooled under vacuum. Under positive nitrogen pressure, the flask is then charged with (S)-1 (1.43 g, 2.91 mmol, 0.1 equiv) and then 20 mL of tetrahydrofuran (THF) is added (Note 8). After the resulting solution is stirred for 30 min, the solvent is removed under vacuum (Note 9). While maintaining a positive flow of pressure, the glass stopper is exchanged for a rubber septum. To the dried ligand is added 80 mL of THF via syringe.
The THF solution of (S)-1 is transferred via cannula into the reaction flask which is maintained at 0 ºC in an ice bath. To ensure that the entire amount of (S)-1 is transferred, an additional 40 mL of THF is added via syringe to the Schlenk flask and then cannula-transferred into the reaction flask. A final 40 mL of THF is added to the round-bottomed flask via syringe, and the reaction mixture is stirred at 0 ºC.
After 1 h, the reaction flask is warmed to room temperature for 15 min. Cyclohexanecarboxaldehyde (3.50 mL, 29.1 mmol, 1 equiv) (Note 10) is then added via syringe. The pale yellow reaction mixture is stirred at room temperature.
After 16 h, the reaction is determined to be complete by TLC (Note 11) and quenched with saturated aqueous ammonium chloride (Note 12). The reaction mixture is then transferred to a 1-L round-bottomed flask, and most of the organic solvent is removed via rotary evaporation (20 ºC, 10 mmHg). The remaining aqueous layer is transferred to a 500-mL separatory funnel and diluted with 200 mL of diethyl ether and an additional 75 mL of saturated aqueous ammonium chloride. After shaking, the organic layer is collected, and the aqueous layer is extracted with ether (3 × 50 mL). The combined organic layers are then washed with water (3 × 100 mL). The organic layer is dried over sodium sulfate (15 g) (Note 13), filtered, and concentrated by rotary evaporation (20 ºC, 10 mmHg to give a non-viscous yellow oil.
This oil is loaded on a coarse-fritted column (5 × 25.4 cm) of SiO2 (170 g) (Note 14). The round-bottomed flask is then rinsed with methylene chloride (10 mL) to ensure that all of the extract is loaded onto the column. The column is eluted as follows: hexanes (1 L), 3% EtOAc/hexanes (1 L), 4% EtOAc/hexanes (500 mL), 5% EtOAc/hexanes (3.5 L). Fraction collection (25 mL fractions) is begun after addition of the second 500 mL portion of 3% EtOAc/hexanes. The product begins to elute at 4% ethyl acetate/hexanes as determined by TLC analysis, and collection is continued until the product is no longer visible by TLC. Due to tailing the product eluted over 3 liters of solvent. Consequently, after 25 fractions of 25 mL that contained product are collected, 500 mL fractions are next collected until no more product is eluted out (Note 15). The desired product is concentrated by rotary evaporation (20 ºC, 10 mmHg) and dried overnight under vacuum (5 × 10-3 mmHg) to yield 5.91 g (92%) of (R)-(+)-α-cyclohexyl-3-methoxybenzenemethanol as a white solid (mp 62-65 ºC) (Notes 16 and 17). HPLC analysis demonstrates that the product is obtained with >99% ee and >99% purity (Note 18).
2. Notes
1.
A nitrogen-filled Vacuum Atmospheres inert atmosphere glove box was used.
2.
Lithium acetylacetonate (97%) was used as purchased from Sigma-Aldrich and stored in the glove box.
3.
To place the flask under nitrogen atmosphere the flask is evacuated (5 × 10
-3 mmHg) and refilled with nitrogen three times. It is crucial that the flask be under a completely inert atmosphere since trace amounts of moisture significantly decrease the yield of product.
4.
1-Methyl-2-pyrrolidinone (99+%, HPLC grade) was purchased from Sigma-Aldrich and refluxed over calcium hydride for 16 h under a nitrogen atmosphere. NMP was then distilled under vacuum (5 × 10
-3 mmHg) at 70-80 ºC. NMP was stored over activated molecular sieves (4 Å) under a nitrogen atmosphere in a dried Schlenk flask (see
Note 3) prior to use.
5.
3-Iodoanisole (99%) was purchased from Sigma-Aldrich and was purified by distillation prior to use. Due to the light sensitivity of 3-iodoanisole, the purification is performed with the careful exclusion of light. 3-Iodoanisole was stirred over activated molecular sieves (4 Å) and copper as a stabilizer (used as received from supplier) for 2 h at 40 ºC. It was then vacuum distilled (5 × 10
-3 mmHg) at 70 ºC and stored in a dried pear-shaped flask under nitrogen atmosphere in the dark.
6.
Diethylzinc (min. 95%) was used as purchased from Strem Chemicals. Diethylzinc is flammable when it comes into contact with water or oxygen in the air and should be handled with caution.
7.
See the preceding procedure for the synthesis of (
S)-
1 [(
S)-3,3'-Bis-morpholinomethyl-5,5',6,6',7,7',8,8'-octahydro-1,1'-bi-2-naphthol].
8.
THF (Certified) was purchased from Fisher Chemicals and distilled under nitrogen atmosphere from sodium benzophenone ketyl.
9.
Under vacuum (5 × 10
-3 mmHg), the flask was warmed with hands and then refilled with nitrogen. This cycle was conducted three times. The flask was then placed under vacuum for 2 h at 40 ºC in an oil bath.
10.
Cyclohexanecarboxaldehyde (97%) was purchased from Sigma-Aldrich and purified prior to use. Cyclohexanecarboxaldehyde was distilled under inert atmosphere at reduced pressure (20 mmHg) from activated molecular sieves (4 Å) directly into a Schlenk tube and sealed. By
1H and
13C NMR, the aldehyde remained pure indefinitely when stored in a Schlenk tube under inert atmosphere.
11.
Aluminum-backed silica gel 60 F
254 TLC plates purchased from EMD Chemicals Inc. were used and visualized with UV light and phosphomolybdic acid (PMA) stain. Hexanes/ethyl acetate (4/1) were used as the eluent. The aldehyde starting material has an R
f = 0.49, and the product has an R
f = 0.24 and is also visible under UV light.
12.
Quenching is an exothermic process and should be performed carefully. Saturated aqueous ammonium chloride was added via syringe through the septum with needles for venting. Over 25 min, 5 mL of saturated aqueous ammonium chloride was slowly added. An additional 20 mL of saturated aqueous ammonium chloride was added over 20 min, using 1 mL aliquots and stirring for 1 min prior to the next addition.
13.
Sodium sulfate (anhydrous) was purchased from Fisher Chemical.
14.
SiliaFlash P60, 40-63 μm 60 Å silica gel was used as purchased from SiliCycle, Inc.
15.
TLC analysis was performed using alumina backed TLC plates
(Note 11), and using hexanes/ethyl acetate (4/1) as eluent. The product was visualized by PMA stain. While the product is UV visible at higher concentrations, PMA stain is necessary to visualize the product at the beginning and end of elution from the column.
16.
(
R)-(+)-
α-Cyclohexyl-3-methoxy-benzenemethanol gave the following analytical data: [a]
D20 +24.7 (
c 1.30, CHCl
3);
1H NMR
pdf (500 MHz CDCl
3) δ: 0.98-1.27 (m, 5 H), 1.42 (apparent d,
J = 13.0 Hz, 1 H), 1.61-1.68 (m, 3 H), 1.79 (m, 1 H), 1.86 (apparent d,
J = 3.5 Hz, 1 H), 2.01 (apparent d,
J = 13.0 Hz, 1 H), 3.85 (s, 3 H), 4.37 (dd,
J = 3.0, 7.0 Hz, 1 H), 6.84 (m, 1 H), 6.90-6.92 (m, 2 H), 7.27 (t,
J = 8.0 Hz, 1 H);
13C
pdf (125 MHz, CDCl
3) δ: 26.0, 26.1, 26.4, 28.8, 29.4, 44.9, 55.2, 79.3, 112.2, 112.8, 119.1, 129.2, 145.4, 159.6; Anal. calcd. for C
14H
20O
2: C, 76.33; H, 9.15; found: C, 76.47; H, 9.49.
17.
The specific rotation was measured on a Perkin-Elmer 241 polarimeter with a sodium lamp (589 nm, D line) using a 1 dm, 2 mL quartz sample cell.
18.
An Agilent 1100 Series Instrument with a multiple-wavelength detector was used for HPLC analysis for the determination of enantiomeric excess and product purity. A Daicel Chiralcel OD column was eluted with 5% isopropanol in hexanes at 1.0 mL/min. The enantiomeric purity was determined to be >99%, and the retention times of the
R- and
S-enantiomers were as follows:
t(S)-1 = 14.6 min, and
t(R)-1 = 23.8 min. The purity was determined to be >99%. The configuration of the product is assigned to be
R as determined by the preparation and NMR analysis of the (
R)- and (
S)-α-methoxy-α-phenylacetic (MPA) ester derivatives of the alcohol product.
11
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Diaryl- and arylalkyl carbinols (
A) are useful intermediates for the synthesis of a number of pharmacologically active compounds.
2,3 Two general strategies for the preparation of these secondary chiral alcohols are the asymmetric reduction of prochiral ketones
4 and the asymmetric arylation of aldehydes.
5 Though both strategies can be carried out catalytically, the latter process generates compounds
A more efficiently because the carbon-carbon bond is formed and the stereocenter is set simultaneously.
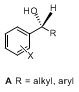
The use of diphenylzinc as the arylating agent has been extensively studied.
6 Because diphenylzinc is the only commercially available diarylzinc reagent, other aryl sources have been explored.
7 For example, preparation of diarylzincs from aryl bromides and
nBuLi at low temperatures and their subsequent asymmetric addition to aldehydes have been studied.
8 Because an alkyllithium is used to generate the arylzincs, the method potentially limits the compatibility of functional groups on the aryl halides.
We have recently reported a catalytic asymmetric method that uses the in situ generation of functionalized arylzincs from aryl iodides for reaction with aldehydes in the presence of the chiral ligand (
S)-
1 (Scheme 1).
9,10 This ligand not only activates the functionalized arylzinc for reaction with aldehydes but also provides excellent stereocontrol. High enantioselectivity has been achieved in the preparation of various diaryl and arylalkyl carbinols by using this method.
9Scheme 1. Preparation of functionalized arylzincs and their addition to aldehydes.
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
Lithium acetylacetonate: 2,4-Pentanedione, ion(1-), lithium (1:1); (18115-70-3)
3-Iodoanisole: Benzene, 1-iodo-3-methoxy-; (766-85-5)
Diethylzinc; (557-20-0)
(S)-3,3'-Bis-morpholinomethyl-5,5',6,6',7,7',8,8'-octahydro-1,1'-bi-2-naphthol: [1,1'-Binaphthalene]-2,2'-diol, 5,5',6,6',7,7',8,8'-octahydro-3,3'-bis(4-morpholinylmethyl)-, (1S)-; (758698-16-7)
Cyclohexanecarboxaldehyde; (2043-61-0)
(R)-(+)-α-Cyclohexyl-3-methoxy-benzenemethanol; (1036645-45-0)
 |
Lin Pu was born in 1965 in Xuyong, Sichuan, China. He received a B.S. degree from Sichuan University in 1984. He then obtained the Doering Fellowship (CGP) and received a Ph.D. degree in 1990 under the supervision of Professor Joseph O'Connor at UC San Diego. From 1991 to 1994, he worked with Professor Henry Taube at Stanford University and Professor Robert Grubbs at California Institute of Technology as a postdoctoral fellow. He was appointed as an assistant professor at North Dakota State University in 1994. He then moved to University of Virginia in 1997 as an associate professor and became a professor in 2003. |
 |
Albert DeBerardinis was born in 1979 and grew up in Aston, Pennsylvania. Encouraged by his entire family, he attended West Chester University from 2000 to 2004, earning a B.S. degree in biochemistry. His undergraduate mentors were invaluable to his interest and further pursuit in chemistry. He was introduced to biochemical research by Professors Blaise Frost and Albert Caffo. Under the direction of Professor Michael Moran, he performed intercalation studies on graphite and prepared polymeric sulfur nitride. After graduating in 2004, he began his doctoral pursuit under the guidance of Professor Lin Pu, working on the development of asymmetric catalysts and their application in organic synthesis. |
 |
Mark Turlington was born in 1984 and grew up in Mills River, North Carolina. In 2002 he attended Furman University and began working in the lab of Professor Moses Lee, synthesizing distamycin analogues capable of molecular recognition of specific DNA sequences. After graduating with a B.S. degree in chemistry in 2006, he is now pursuing his doctoral degree under the guidance of Professor Lin Pu, working on the development of asymmetric catalysts and their application in organic synthesis. |
 |
Kyle Kimmel was born in 1985 in Fairfax, VA. He studied as an undergraduate at the University of California, Irvine, where he completed a B.S. degree in Chemistry, working on undergraduate research in the lab of Professor Keith Woerpel. Currently, he is a third year graduate student at University of California, Berkeley, working under the direction of Professor Jonathan A. Ellman. His research is on the development of sulfinyl-based hydrogen-bonding organocatalysts. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved