Org. Synth. 2010, 87, 253
DOI: 10.15227/orgsyn.087.0253
PREPARATION AND [2+2] CYCLOADDITION OF 1-TRIISOPROPYLSILOXY-1-HEXYNE WITH METHYL CROTONATE: 3-BUTYL-4-METHYL-2-TRIISOPROPYLSILOXY-CYCLOBUT-2-ENECARBOXYLIC ACID METHYL ESTER
Submitted by Valeriy Shubinets, Michael P. Schramm, and Sergey A. Kozmin
1.
Checked by John Frederick B. Briones and Huw M. L. Davies.
1. Procedure
Caution! Reactions and subsequent operations involving peracids and peroxy compounds should be run behind a safety shield. Peroxy compounds should be added to the organic material, never the reverse. For relatively fast reactions, the rate of addition of the peroxy compound should be slow enough so that it reacts rapidly and no significant unreacted excess is allowed to build up. The reaction mixture should be stirred efficiently while the peroxy compound is being added, and cooling should generally be provided since many reactions of peroxy compounds are exothermic. New or unfamiliar reactions, particularly those run at elevated temperatures, should be run first on a small scale. Reaction products should never be recovered from the final reaction mixture by distillation until all residual active oxygen compounds (including unreacted peroxy compounds) have been destroyed. Decomposition of active oxygen compounds may be accomplished by the procedure described in Korach, M.; Nielsen, D. R.; Rideout, W. H. Org. Synth. 1962, 42, 50 (Org. Synth. 1973, Coll. Vol. 5, 414). [Note added April 2018].
A. 1-Triisopropylsiloxy-1-hexyne (2). A three-necked, flame-dried, 500-mL, round-bottomed flask equipped with an egg-shaped 1 1/4 × 5/8 in magnetic stir bar, rubber septum fitted with argon inlet needle, and rubber stopper fitted with thermometer is flushed with argon and charged with THF (100
mL) (Note 1) and 1-hexyne (3.44 mL, 30 mmol, 1.0 equiv) (Note 2) by syringe through a septum. The solution is cooled in a dry ice acetone bath to -78 °C and treated dropwise over 10 min with anhydrous t-BuOOH (6.30 mL of 5.26 M solution in nonane, 33 mmol, 1.1 equiv) (Note 3) by means of a syringe pump (Note 4). The resulting mixture is treated dropwise with lithium bis(trimethylsilyl)-amide (69 mL of 1.0 M solution in THF, 69 mmol, 2.3 equiv) (Note 5) over 30 min by means of a syringe pump (Note 6). The flask is then placed in a 0 °C ice water bath and allowed to stir for 2 h. The reaction mixture is cooled in a dry ice acetone bath to -78 °C and treated dropwise over 10 min with triisopropylsilyl trifluoromethanesulfonate (8.90 mL, 33 mmol, 1.1 equiv) (Note 7) by means of a syringe pump (Note 8). The reaction mixture is stirred for 5 min, then placed in a 0 °C ice water bath and allowed to stir for 40 min. The reaction mixture is then diluted with hexanes (200
mL) (Note 9) and transferred into a 1-L separatory funnel containing saturated aqueous NaHCO3 solution (125
mL), using an additional 100 mL of hexanes to wash the flask. The layers are separated. The aqueous layer is extracted with hexanes (75). The combined organic layers are washed with saturated aqueous Na2S2O3 (100
mL) (Note 10), 100 mL of brine, and dried over anhydrous MgSO4. The solution is filtered through a fritted glass funnel with fine porosity. The solvent is removed on a rotary evaporator (Note 11). The crude product is transferred to a 50-mL round-bottomed flask using two hexanes washings. Hexane solvent is removed using a rotary evaporator, and the product is purified by fractional distillation (Notes 12 and 13) to afford 6.66 g (87%) of 1-triisopropylsiloxy-1-hexyne 2 (Note 14).
B. 3-Butyl-4-methyl-2-triisopropylsilanyloxy-cyclobut-2-enecarboxylicacid methyl ester (
4). A single-necked, dry, 250-mL, round-bottomed flask equipped with an egg-shaped 1 × 1/2 in. magnetic stirbar is charged with
1-triisopropylsiloxy-1-hexyne 2 (5.12 g, 20.16 mmol, 1.2 equiv),
dry methylene chloride (140
mL) (Note 15), and
methyl crotonate (1.78 mL, 16.8 mmol, 1.0 equiv) (Note 16) at 23 °C.
Silver trifluoromethylsulfonimide4 (326 mg, 0.84 mmol, 0.05 equiv) (Note 17) is added in one portion and the reaction mixture is stirred for 15 min at which time TLC
(Note 18) indicates complete consumption of starting material and the product formation is evident from a new spot with R
f = 0.53 (10:1 hexanes:ether), corresponding to siloxy cyclobutene
4. The reaction mixture is washed with
brine (50
mL). The aqueous phase is then extracted with
methylene chloride (25
mL). The combined organic layers are dried over anhydrous MgSO
4 and filtered through a fritted glass funnel with fine porosity. The solvent is removed on a rotary evaporator and the crude product is purified by column chromatography
(Note 19) using 85 g of SiO
2 (elution with 10:1, hexanes:ether). The fractions containing the product are concentrated to afford 4.57-4.79 g (77-80% yield) of 3-butyl-4-methyl-2-triisopropylsilanyloxy-cyclobut-2-enecarboxylicacid methyl ester
4as a colorless oil
(Note 20).
2. Notes
1.
THF (HPLC grade, H
2O = 0.003%) was purchased from Fisher Scientific Company and distilled from sodium benzophenone ketyl.
2.
1-Hexyne (97%) was purchased from Aldrich Chemical Company, Inc. and used without further purification.
3.
The solution of t-BuOOH (5.26 M in nonane) was purchased from Aldrich Chemical Company, Inc. (Fluka). It was sold as an approximately 5.5 M solution, but the exact concentration (up to one-hundredth) of a particular batch can be obtained at the Aldrich website.
4.
During the addition of t-BuOOH the internal temperature was maintained between -65 and -60 °C.
5.
The solution of lithium bis(trimethylsilyl)-amide (1.0 M in THF) was purchased from Aldrich Chemical Company, Inc.
6.
During the addition of LiHMDS, the internal temperature was kept steady at -60 °C.
7.
Triisopropylsilyl trifluoromethanesulfonate (97%) was purchased from Aldrich Chemical Company, Inc. and fractionally distilled over calcium hydride at 3 mmHg.
8.
During the addition of TIPSOTf, the internal temperature was maintained between -65 and -60 °C
9.
Hexanes (ACS grade) were purchased from Fisher Scientific Company and used without further purification.
10.
This operation is intended to remove any residual t-BuOOH. In the past, however, when this operation was omitted from the procedure, the NMR of the crude product did not show any presence of t-BuOOH. Nevertheless, washing the organic phase with saturated aqueous Na
2S
2O
3 is recommended as a safety precaution.
11.
Most of the nonane can be removed on a rotary evaporator at 40 °C using a diaphragm pump (ca. 20 mmHg).
12.
Fractional distillation was done using a vigreux column. 1-Triisopropylsiloxy-1-hexyne was distilled at 102-105 °C (ca. 3 mmHg). Care must be taken at the beginning of the distillation process as the crude siloxy alkyne has a tendency to foam when placed under high vacuum (most likely as a result of the leftover nonane). A small amount of triisopropyl silanol (ca. 0.3-0.4 mL) is removed at 70-90 °C.
13.
Alternatively, one could distill the siloxy alkyne using bulb-to-bulb distillation (first removing the triisopropyl silanol and then distill the siloxy alkyne itself).
14.
Occasionally, a small amount of impurities (such as triisopropyl silanol) may be observed. The exact amount of these impurities depends on how well the distillation is performed. 1-Triisopropylsiloxy-1-hexyne
2displays the following spectral data: IR (neat) 2945, 2869, 2278, 1463, 1254 cm
-1;
1H NMR
pdf (400 MHz, C
6D
6) δ: 0.77 (t, 3 H,
J= 6.8 Hz), 1.01-1.04 (m, 18 H), 1.07-1.13 (m, 3 H), 1.33-1.38 (m, 4 H), 2.06 (t, 2 H,
J = 7.2 Hz);
13C NMR
pdf (100 MHz, C
6D
6) δ: 12.0, 13.6, 17.2, 17.3, 22.1, 30.7, 32.4, 87.1; MS (APCI)
m/z calcd. for C
15H
31OSi ([M+H]
+) 255.2139; found 255.2144; Anal. calcd. for C
15H
30OSi: C, 70.79; H, 11.88; found: C, 70.49; H, 11.97.
15.
Methylene chloride (HPLC grade) was purchased from Fisher Scientific Company and distilled over calcium hydride.
16.
Methyl crotonate (98%) was purchased from Aldrich Chemical Company, Inc. and used without further purification.
17.
Silver trifluoromethylsulfonimide was prepared according to a literature procedure:
4 Trifluoromethanesulfonimide (2.81 g, 10 mmol, 95% from Aldrich Chemical Company, Inc.) was added to a 100-mL round-bottomed flask, which was wrapped in aluminum foil. To this flask, 20 mL of distilled water was added, followed by
silver carbonate (1.5 g, 5.4 mmol, 99% from Aldrich Chemical Company, Inc.). The flask was warmed to 65 °C in an oil bath and stirred for 4 h, after which the water was removed using a rotary evaporator (10 mmHg). The flask was placed on a vacuum line (1 mmHg) and heated to 80 °C for 14 h. It was removed from the vacuum line, brought to room temperature, and treated with 30 mL of diethyl ether (anhydrous grade, Fisher Scientific Company), stirred for 2 h, and filtered through a glass sintered frit (fine grade) to remove some small black impurities. The filtrate was concentrated on a rotary evaporator to give a powder. The powder was placed under vacuum (1 mmHg) at room temperature for 12 h to remove any remaining water and ether to give 3.31 g of silver trifluoromethylsulfonimide (85 % yield).
18.
TLC was performed using Whatman precoated 60 Å silica gel plates with fluorescent indicator.
19.
Flash column chromatography was performed over Silicycles Inc. ultra pure 230-400 mesh silica gel, pH 7.0, H
2O content = 6%. Fractions of ca 15 mL each were collected using 16 × 150 mm test tubes. A total of 26 fractions were collected and fractions 12 to 18 were pooled and concentrated to give the desired product.
20.
Cyclobutene
4 displays the following spectral data: IR (neat) 2947, 2867, 1737, 1702 cm
-1;
1H NMR
pdf (400 MHz, CDCl
3) δ: 0.86 (t, 3 H,
J = 7.2 Hz), 1.02-1.07 (m, 21 H), 1.10 (d, 3 H,
J = 6.8 Hz), 1.20-1.40 (m, 4 H), 1.85-1.92 (m, 1 H), 2.01-2.09 (m, 1 H), 2.41 (q, 1 H,
J = 6.8 Hz), 3.00 (br s, 1 H), 3.63 (s, 3 H);
13C NMR
pdf (100 MHz, CDCl
3) δ: 12.8, 14.0, 17.80, 17.9, 18.1, 22.9, 24.8, 29.4, 35.1, 51.7, 56.6, 125.6, 138.6, 173.6; MS (APCl)
m/z calcd. for C
20H
39O
3Si ([M+H]
+) 355.2663; found 355.2670; Anal. calcd. for C
20H
38O
3Si: C, 67.74; H, 10.80; found: C, 67.44; H, 10.87.
Safety and Waste Disposal Information All hazardous materials should be handled and disposed of in accordance with "Prudent Practices in the Laboratory"; National Academy Press; Washington, DC, 1995.
3. Discussion
Siloxy alkynes represent a general class of organosilicon compounds with significant potential for the development of an arsenal of new synthetic methods having both practical and conceptual untility in organic synthesis.
3,5-9 Among several existing methods for preparation of siloxy alkynes, the protocol developed by Julia in 1993
2 represents perhaps the most simple, versatile and efficient approach to these compounds. The Julia method entails the generation of an acetylide anion from a terminal alkyne, followed by addition of lithium tert-butyl hydroperoxide, which at 0 °C triggers a facile oxidation of the lithium acetylide to give lithium ynolate and lithium tert-butoxide. The modification of the original procedure reported herein is based on the use of triisopropylsilyl triflate instead of the silyl chloride, which was utilized in the initial report by Julia. This modification enables the highly efficient silylation of the ynolate anion to furnish the corresponding siloxy alkyne in excellent yield. Importanly, only 1 equivalent of silyl trilfate is sufficient for the silylation step. Presumably, the competitive silylation of lithium tert-butoxide is significantly slower.
Scheme 1
This simple protocol was utilized to convert a range of terminal alkynes to the corresponding 1-siloxy-1-alkynes. Several representative examples are depicted in Table 1. We found that the use of bulky siloxy groups, i.e. TIPS, was required to improve the hydrolytic stability of the resulting alkynes, which can be readily purified via distillation or chromatography on SiO2. In many instances, the alkynes were obtained in sufficiently pure form for direct use in the next transformation.
Table 1 Yields of various 1-siloxy-1-alkynes
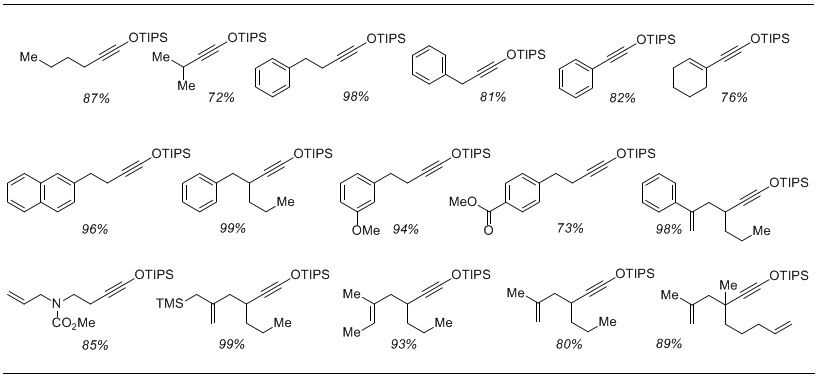
We found that a catalytic amount of silver trifluoromethylsulfonamide can efficiently promote the [2+2] cycloadditions of siloxy alkynes with simple unsaturated ketones, esters, and nitriles.
3 This transformation provided an efficient method for the assembly of a range of highly functionalized siloxy cyclobutenes. The investigation of the scope of this method revealed that many unsaturated carbonyl compounds are capable of efficient participation in this reaction with a range of siloxy alkynes (Table 2). In most cases, these reactions proceeded to completion in 5 minutes at room temperature, demonstrating the mild and general nature of this catalytic protocol. Two representative examples of subsequent transformations of siloxy cyclobutenes are depicted in Scheme 2. Direct protodesilylation of silyl enol ether afforded keto acid in 95% yield, arising from the fragmentation of the initially produced acyl cyclobutanone. Alternatively, a highly diastereoselective DIBAL reduction (dr 92:8), followed by protodesilylation furnished the cyclobutenone (dr 55:45) without any detectable ring-opening products.
Scheme 2
Table 2 Selected [2+2] cycloadditions of siloxy alkynes
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
Methyl crotonate; (623-43-8)
Triisopropylsilyl trifluoromethanesulfonate; (80522-42-5)
Trifluoromethanesulfonimide; (82113-65-3)
Silver carbonate ; (534-16-7)
 |
Sergey A. Kozmin received his Undergraduate Diploma at the Moscow State University in 1993. He obtained his Ph.D. in 1998 at the University of Chicago with Viresh H. Rawal, and completed postdoctoral studies at the University of Pennsylvania with Amos B. Smith, III in 2000. Kozmin is currently an Associate Professor of Chemistry at the University of Chicago. His research program combines several complementary efforts, including (a) development of new catalytic reactions of conceptual and practical utility; (b) synthesis of complex natural products of notable biological significance; and (c) generation of highly diverse chemical libraries, featuring the complexity of natural metabolites. |
 |
Valeriy Shubinets was born in 1985 in Ivano-Frankivsk, Ukraine. He obtained his B.S. in Chemistry and Biochemistry and his B.A. in Biology in 2007 from the University of Chicago, where he performed his undergraduate research with Professor Sergey Kozmin in the area of new method development and natural product synthesis. Valeriy is currently a student at the Harvard Medical School. |
 |
Michael P. Schramm was born in Syracuse, New York in 1975. He received his Ph.D. under the direction of Professor Sergey A. Kozmin from The University of Chicago in 2005. He then completed two years of postdoctoral work under the supervision of Professor Julius Rebek, Jr. at The Scripps Research Institute in La Jolla, California. In 2007 he began his independent career as Assistant Professor in the Department of Chemistry and Biochemistry at California State University Long Beach. His research efforts utilize principles of molecular recognition to design and prepare modular alpha-helicial peptidomimetic libraries. Additionally, these principles are being directed at the preparation and study of synthetic small molecule membrane transporters. |
 |
John Frederick Briones was born in 1982 in Laguna, Philippines. He earned his B.S. degree in Chemistry from the University of the Philippines, Los Banos in 2003 and later on pursued his Master's degree at the University of the Philippines, Diliman. He joined the research lab of Prof. Huw Davies in 2007 and currently his research project focuses on Rh(II) catalyzed enantioselective cyclopropenation of alkynes. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved