Org. Synth. 2011, 88, 121-137
DOI: 10.15227/orgsyn.088.0121
ORGANOCATALYTIC ENANTIOSELECTIVE SYNTHESIS OF BICYCLIC ß-LACTONES FROM ALDEHYDE ACIDS VIA NUCLEOPHILE-CATALYZED ALDOL-LACTONIZATION (NCAL)
Submitted by Henry Nguyen, Seongho Oh, Huda Henry-Riyad, Diana Sepulveda, and Daniel Romo
1
.
Checked by Gerri E. Hutson and Viresh H. Rawal
2
.
1. Procedure
Caution: Care must be exercised in transferring manipulating the ozonide reaction mixture due to the known instability of ozonides. It is best to perform the ozonide reduction step in the same fume hood avoid transferring manipulation of the reaction mixture.
A.
(1,4-Dioxaspiro[4.5]dec-7-en-8-yloxy)trimethylsilane (
1
)
(Note 1). An oven-dried 500-mL, two-necked, round-bottomed flask is equipped with a large stir bar (Note 2), and one neck is fitted with a 50-mL pressure-equalizing addition funnel, to which is attached a nitrogen inlet, and the second neck is sealed with a rubber septum. The reaction flask is charged with solid
1,4-cyclohexanedione mono-ethylene ketal (25.0 g, 160 mmol, 1.0 equiv), and then N,N-dimethylformamide (Note 3) (100 mL) is added to dissolve the solid.
Triethylamine (52.0 mL, 368 mmol, 2.3 equiv) is added via syringe to the reaction vessel. Freshly distilled
trimethylsilyl chloride (26.7 mL, 208 mmol, 1.3 equiv) is added to the addition funnel and then dispensed dropwise into the reaction vessel over a period of ~15 min. After the addition is complete, the addition funnel is replaced with a condenser, and the reaction mixture is then heated in an oil bath at 80 °C (bath temperature) for 12 h (Note 4). After cooling to ambient temperature (23 °C), the reaction is quenched by addition of cold H2O (150 mL, cooled in an ice bath for 15 min), and the resulting mixture is diluted with
hexanes (300 mL) and then transferred to a 1000-mL separatory funnel. The aqueous layer is removed and back-extracted with
hexanes (2 × 100 mL). The combined hexane extracts are washed with saturated aqueous NaHCO3 (1 × 100 mL), saturated aqueous NH4Cl (2 × 150 mL), and then
brine (1 × 150 mL). The organic layer is dried over anhydrous MgSO4 (25 g) for 15 min and then concentrated on a rotary evaporator (25 mmHg, 30 °C). Further concentration under reduced pressure (0.5 mmHg, ambient temperature, 23 °C) delivers 32.6 g (89%) of the silyl enol ether 1
(Note 5) as a dark yellow oil, which is of sufficient purity for use in the subsequent step.
B.
3-(2-(2-Oxoethyl)-1,3-dioxolan-2-yl) propanoic acid (2).
To a 1000-mL round-bottomed flask fitted with an egg-shapped magnetic stir bar (0.625 × 1.5 in) is added crude
silyl enol ether 1
(32.0 g, 140 mmol, 1.0 equiv) and 500 mL of CH2Cl2. The reaction mixture is cooled to -78 °C (acetone/dry ice bath temperature) with stirring for 15 min. Stirring is stopped and ozone is bubbled through the solution with a fritted-glass gas dispersion tube for ~1-2 h until the blue color of ozone persists (Note 6). A stream of nitrogen is then bubbled through the solution until the blue color disappears (~ 1 h). Solid triphenylphosphine (37.1 g, 141 mmol, 1.01 equiv) is added with stirring, and the reaction mixture is allowed to warm gradually to ambient temperature (23 °C) and stirred for ~18 h until the ozonide test provides a negative result (Note 7). The solvent is removed by means of a rotary evaporator (25 mmHg, 30 °C). Residual CH2Cl2 is removed by dissolving the crude mixture in
diethyl ether (50 mL) and concentrating the solution on a rotary evaporator (25 mmHg, 30 °C). This step is repeated twice, and the resulting yellow slurry is then placed under high vacuum (0.5 mmHg) for 12 h. (Note 8).
Diethyl ether (80 mL) is added, the solid/liquid mixture is stirred vigorously for 30 min, and the solid is then filtered off by vacuum filtration using a Büchner funnel fitted with filter paper (Whatmann #1, 70 mm). The solid is rinsed with ice cold ether (2 × 15 mL). The ether filtrates are combined and transferred to a separatory funnel (250 mL) and extracted vigorously with water (4 × 80 mL) (Note 9). To a 500-mL Erlenmeyer flask containing the combined aqueous extracts (~250 mL) is added solid NaCl (70 g) with stirring, followed by additional NaCl until the solution is saturated. The aqueous solution is transferred to a 50-mL separatory funnel and extracted with
hexanes (2 × 100 mL) to remove unreacted cyclohexane dione monoketal from step A (Note 4). The aqueous solution is then transferred to a clean 1000-mL separatory funnel and extracted vigorously with CH2Cl2 (3 × 150 mL). The aqueous solution is saturated again with additional
sodium chloride (5 g) and extracted vigorously again with CH2Cl2 (3 × 150 mL). The combined CH2Cl2 extracts are dried over anhydrous MgSO4 (50 g) for 15 min, filtered using a Büchner funnel equipped with filter paper (Whatman #1, 70 mm), and concentrated by rotary evaporation (25 mmHg, 30 °C) to deliver a light yellow oil. Further concentration under high vacuum (0.5 mmHg, 23 °C) provides 15.1 g (57%) of the aldehyde acid as a light yellow solid (Note 10), which is of sufficient purity for use in the subsequent step without further purification (Notes 11 and 12).
C.
N-Methyl-2-chloropyridinium trifluoromethane sulfonate (
3).
A 1000-mL round-bottomed flask, equipped with an egg-shapped magnetic stir bar (0.625 × 1.5 in) and a rubber septum, is flame dried under a stream of nitrogen and cooled to ambient temperature. Following the addition of CH2Cl2 (250 mL) and
2-chloropyridine (13.1 mL, 139 mmol, 1.0 equiv), the flask is cooled in a dry ice/acetone bath (-78 °C, bath temperature) under nitrogen.
Methyl trifluoromethanesulfonate (25.0 g, 153 mmol, 1.1 equiv) is then added slowly via syringe over 10 min through the septum down the side of the flask to allow cooling prior to mixing with the bulk solvent. The cold bath is removed and the reaction mixture is allowed to warm slowly to ambient temperature (23 °C) and stirred an additional 12 h, leading to a white precipitate. The reaction mixture is concentrated by rotary evaporation (25 mmHg, 30 °C) to deliver a white solid.
Toluene (120 mL) is added to further induce precipitation, and after swirling under nitrogen for ~10 minutes, the toluene/CH2Cl2 mixture is removed from the solids via cannula under N2 pressure. The white solid is dried under reduced pressure at 0.5 mmHg for 12 h at 23 °C to afford 35.9 g (93%) of
N-methyl-2-chloropyridinium trifluoromethane sulfonate
(3) as a white solid (mp 147-148 °C) (Note 13).
D.
O-Trimethylsilylquinidine (
4
, O-TMS QND)
.
3
A 250-mL round-bottomed flask, equipped with a mechanical stirrer (PTFE stirrer blade 1.9 × 6 cm) is flame-dried with a stream of nitrogen and left to cool down to ambient temperature for 10 min. To the flask are added
quinidine (4.00 g, 12.4 mmol, 1.0 equiv) and CH
2Cl
2 (100 mL) followed by
distilled
trimethylsilyl chloride (1.9 mL, 15 mmol, 1.2 equiv), which is added slowly via syringe pump over 15 min. The resulting solution is stirred at ambient temperature for 24 h and then transferred to a 500-mL separatory funnel, to which was added CH
2Cl
2 (100 mL)
and NaHCO
3 (aq) (100 mL). After shaking, the layers are separated, and the aqueous layer is reextracted with CH
2Cl
2 (2 × 50 mL). The combined organic extracts are dried over MgSO
4, filtered, and concentrated by rotary evaporation (25 mmHg, 30 °C). The residue is then left to dry under reduced pressure at 0.5 mmHg for 12 h at 23 °C to afford 4.02 g (82%) of
O-trimethylsilylquinidine (4)
as a clear oil, which was of sufficient purity for use without purification
(Note 14).
E.
(1'S,5'R)-Spiro[1,3-dioxolane-2,3'-[6]oxabicyclo[3.2.0]heptan]-7'-one (
5
).
N-Methyl-2-chloropyridinium trifluoromethane sulfonate (3)
(Note 13) (33.2 g, 120 mmol, 1.5 equiv) is weighed out in a one-necked, oven-dried 1000-mL round-bottomed flask. A 4-cm stir bar is added, and the flask is purged with nitrogen and then charged with dry
acetonitrile (240 mL). A solution of the catalyst, O-TMS QND (4) (3.16 g, 7.97 mmol, 0.1 equiv) dissolved in dry
acetonitrile (100 mL) at 23 °C is transferred to the reaction flask via cannula with nitrogen pressure, which is followed by addition of freshly distilled diisopropylethylamine (Hünig's base) via syringe (35.4 mL, 199 mmol, 2.5 equiv). The reaction mixture is stirred for 10 min, and then a solution of
3-(2-(2-oxoethyl)-1,3-dioxolan-2-yl) propanoic acid (2) (15.0 g, 79.7 mmol, 1.0 equiv) in dry CH3CN (60 mL) is added via syringe pump over ~1 h (30 mL syringe, ~1 mL/min, syringe is loaded twice). The reaction mixture changes from yellow to a dark red color during this addition. The reaction mixture is stirred for an additional 18 h at ambient temperature, and the reaction progress is monitored by the disappearance of aldehyde acid 2 and the formation of β-lactone 5 by TLC analysis (R
f
0.29 and 0.52, respectively in 60% EtOAc/hexanes, KMnO4 stain). When the reaction is judged complete, the reaction mixture is concentrated by rotary evaporation and then by high vacuum (0.5 mmHg, 30 °C) for 3 h to provide a dark brown, viscous oil (~ 75 g). The oil is diluted with 25 mL of CH2Cl2 and loaded onto a silica gel plug (188 g, 2.5 g silica gel per gram of crude product, 10-cm diameter flash column) pre-eluted with 50% EtOAc:hexanes, using a long stem funnel to carefully deliver the crude mixture onto the silica gel surface (washing with 5 mL of CH2Cl2 and then 10 mL of 50% EtOAc:hexanes). The product is quickly eluted with 50%
EtOAc:hexanes (2000 mL). Forty fractions (~45 mL in 50 mL test tubes) are collected and fractions 7-35 containing the majority of the β-lactone (R
f
= 0.52 (60% EtOAc:hexanes; KMnO4)) are combined and concentrated by rotary evaporation (25 mmHg, 30 °C) and then under high vacuum (0.5 mmHg at 23 °C) to deliver 8.3 g of semi-crude β-lactone. Recrystallization (Note 15) from EtOAc:hexanes gives a total of 4.5 g (33%) of bicyclic-β-lactone (Notes 16 and 17). Enantiomeric excess was determined to be 90% ee by chiral HPLC analysis (Note 18).
2. Notes
1.
Sources and purities of reagents used in the procedure are as indicated: 1,4-Cyclohexanedione mono-ethylene ketal (Aldrich, 97%), PPh
3 (Acros, 99%), 2-chloropyridine (Aldrich, 99%), methyl trifluoromethanesulfonate (Acros, 96%), quinidine (Acros, 99%), NEt
3 (Acros, 99%), TMSCl (Acros, 98%),
N,N-dimethylformamide (Aldrich, 99.9%), acetonitrile (Aldrich, 99.8%),
N, N-diisopropylethyl amine (Hünig's base, Acros, 98%), MgSO
4 (EMD, powder 98%), sodium bicarbonate (EMD, powder), dichloromethane (EMD, 99.8%), NaCl (EMD, crystals, 99%), ammonium chloride (EMD, 99.5%), hexanes (Fisher, 98.5%), diethylether (EMD, 99%), silica gel (Silicycle, 230-400 mesh), KI (EMD, 99%). Triethylamine, trimethylsilyl chloride, and Hünig's base were freshly distilled over CaH
2 prior to use.
N,N-Dimethylformamide, dichloromethane and acetonitrile were dried through activated alumina using a converted MBraun System. Checkers used a solvent drying system manufactured by Innovative Technology, Inc.
2.
A 4-cm football-shaped stir bar is ideal since copious amounts of Et
3N•HCl formed during the course of the reaction makes stirring difficult.
3.
It is important to use very dry
N,
N-dimethylformamide (<150 ppm of H
2O) to avoid hydrolysis of TMSCl.
4.
While it is best to have all starting material converted to the silyl enol ether, reaction progress is not easily monitored by TLC analysis due to facile desilylation of the TMS enol ether
1. Taking an aliquot for NMR analysis is appropriate. Generally, ensuring high quality and dryness of reagents/solvents and using specified reaction times ensured complete reaction. Any residual cyclohexanone is not deleterious to the subsequent ozonolysis and can be removed in the next step during extraction.
5.
The checkers obtained 17.03 g (93%) on a one-half scale run. The submitters reported obtaining 31-33 g (85-90%) of
1. Data for silyl enol ether
1: R
f
= 0.65 (20% EtOAc/hexanes); IR (thin film) 1670 cm
−1;
1H NMR
pdf (500 MHz, CDCl
3) δ: 0.17 (s, 9 H), 1.78 (t,
J = 6.5 Hz, 2 H), 2.16-2.27 (m, 4 H), 3.93-3.98 (m, 4 H), 4.68-4.73 (m, 1 H)
;
13C
pdf (125 MHz, CDCl
3) δ: 0.27, 28.5, 31.1, 33.9, 64.3, 100.6, 107.6, 149.8
; [M + Na
+] calcd for C
11H
20O
3SiNa
+: 251.10739. Found: 251.10743.
6.
A Welbash Ozonizer (total pressure: 7-10 psi; O
2/ozone pressure: 3.0-4.5 psi) was used by the submitters and the checkers. The time required for the ozonolysis is dependent upon the O
2 pressure and ozone generation efficiency.
7.
The ozonide test is performed by adding 0.1 g of potassium iodide to 1.0 mL of glacial acetic acid and ~1.0 mL of the ozonolysis reaction mixture in a 5 mL round-bottomed flask. A brown mixture indicates the presence of ozonide. Blanks must always be prepared: 0.1 g of KI is added to 1.0 mL of CH
2Cl
2 and 1.0 mL of glacial acetic acid. The test solution has a very short shelf life and will naturally result in high blank values if stored for any length of time.
Caution: Care must be exercised in transferring and manipulating the ozonide reaction mixture due to the known instability of ozonides. It is preferable to perform the quenching step in the same fume hood and avoid transferring the reaction mixture. A test for the presence of ozone resulting in a yellow solution, indicative of the absence of peroxides, is required before it is safe to proceed to work up.
8.
The CH
2Cl
2 must be removed at this step to minimize retention of aldehyde acid
2 in the organic layer during the first stage of the extraction procedure.
9.
It is imperative that extractions of the aldehyde acid are done vigorously, with rapid end-to-end shaking, to ensure efficient transfer of the product to the aqueous phase. However, patience is then required to allow for separation of the layers due to emulsion formation. A copper wire loop was used by the submitters to assist in breakdown of the emulsion.
10.
The submitters reported 57-65% yield of product
2 on full scale.
11.
It is best to use the aldehyde acid soon after drying otherwise it should be stored in the freezer (-5 °C) under N
2 to avoid oxidation to the diacid. Data for aldehyde acid: R
f
= 0.29 (60% EtOAc:hexanes); IR (thin film) 1723 cm
−1;
1H NMR
pdf(500 MHz, CDCl
3) δ: 2.11 (t,
J = 7.5 Hz, 2 H), 2.45 (t,
J = 7.5 Hz, 2 H), 2.69 (d,
J = 3.0 Hz, 2 H), 4.03 (s, 4 H), 9.73 (t,
J = 3.0 Hz, 1 H)
;
13C
pdf (125 MHz, CDCl
3) δ: 28.2, 33.0, 50.6, 65.2, 108.3, 178.9, 199.8
; [M - H] calcd for C
8H
11O
5: 187.0612. Found: 187.0612.
12.
A minor product formed during the ozonolysis was identified to be the corresponding diacid
4 formed by over oxidation and this is removed during the extraction procedure.
13.
The pyridinium salt is somewhat hygroscopic and moisture sensitive and thus should be weighed out and transferred rapidly. IR (film) 3099, 1619, 1264 cm
−1;
1H NMR
pdf (500 MHz, CD
3CN) δ: 4.29 (s, 3 H), 7.93-7.96 (m, 1 H), 8.12-8.14 (m, 1 H), 8.45-8.49 (m, 1 H), 8.78-8.80 (m, 1 H)
;
13C NMR
pdf (125 MHz, CD
3CN) δ: 48.9, 122.3, 126.6, 127.6, 131.2, 148.6, 149.3
.
14.
Characterization data: IR (thin film) 2939, 1621, 1507 cm
−1; This compound is a mixture of two conformers at ambient temperature but low temperature
1H NMR at -40 °C allowed assignment of one major conformer
pdf (500 MHz, -40 °C, CDCl
3) δ: 0.15 (s, 9 H), 0.75-0.89 (m, 1 H), 1.31-1.53 (m, 2 H), 1.68 (br s, 1 H), 2.11 (t,
J = 11.5 Hz, 1 H), 2.15-2.25 (m, 1 H), 2.76-2.98 (m, 4 H), 3.38 (dd,
J = 8.0, 12.0 Hz, 1 H), 3.92 (s, 3 H), 5.03 (d,
J = 9.0 Hz, 1 H), 5.05 (d,
J = 17.5 Hz, 1 H), 5.69 (br s, 1 H), 6.08 (apparent quint,
J = 9.0 Hz, 1 H), 7.08 (br s, 1 H), 7.36 (dd,
J = 2.5, 9.0 Hz, 1 H), 7.53 (d,
J = 4.5 Hz, 1 H), 8.01 (d,
J = 9.0 Hz, 1 H), 8.72 (d,
J = 4.5 Hz, 1 H)
;
13C NMR was taken at ambient temperature.
13C NMR
pdf (125 MHz, CDCl
3) δ: 0.1, 19.8, 26.5, 28.2, 40.3, 49.7, 50.5, 55.6, 60.3, 73.3, 100.4, 114.3, 118.5, 121.5, 126.1, 131.8, 140.8, 144.3, 147.4, 147.6, 157.8
.
15.
Recrystallization is performed in a 50-mL Erlenmeyer flask by addition of hot
EtOAc (20 mL) to the dried, semi-crude β-lactone. A stir bar is added, and the solution is then heated with stirring to ~50 °C on a hot plate to dissolve the solid.
Hexanes (~16 mL) is added to the hot solution with continued heating until the solution turns cloudy. On cooling to ambient temperature over ~1 h, crystals form and the flask is then placed in an ice-bath for an additional 30 min. The crystals are collected by rapid vacuum filtration and washed quickly with an ice-cold mixture of 50%
EtOAc:hexanes (~10 mL) to give 4.0-4.2 g (30-31%) of the bicyclic-β-lactone as light yellow crystals. A second crop is collected by removal of ~1/3 of the mother liquor and placing the solution in a freezer (-10 °C) overnight to provide an additional 0.5-0.6 g (4-5%) as darker yellow crystals.
16.
The submitters reported 34-35% yield of product
5 on full scale.
17.
Higher yields of β-lactone could be achieved by using aldehyde acid purified by flash chromatography; however; a loss of material on the column led to further reduction of the overall yield. Characterization data for (1'
S,5'
R)-spiro[1,3-dioxolane-2,3'-[6]oxabicyclo[3.2.0]heptan]-7'-one (
5): R
f
= 0.52 (60% EtOAc/hexanes); mp 103-104 °C; [α]
D
23 + 63.2 (
c 0.95, CHCl
3); IR (thin film) 1825 cm
−1;
1H NMR
pdf (500 MHz, CDCl
3) δ: 2.01 (dd,
J = 8.5, 14.0 Hz, 1 H), 2.06 (dd,
J = 5.0, 15.5 Hz, 1 H), 2.29 (dd,
J = 1.0, 14.0 Hz, 1 H), 2.36 (dd,
J = 1.5, 15.5 Hz, 1 H), 3.85-3.92 (m, 2 H), 3.97-4.07 (m, 3 H), 4.99 (t,
J = 5.0 Hz, 1 H)
;
13C NMR
pdf (125 MHz, CDCl
3) δ: 36.6, 39.5, 53.6, 64.8, 65.0, 74.0, 115.3, 170.6
; [M + Na
+] calcd for C
8H
10O
4Na
+: 193.0471. Found: 193.0476.
18.
Enantiomeric excess was determined to be 90% ee by chiral HPLC using the following conditions: Daicel IA column, 90% hexanes/10% isopropanol, 1 mL/min, 25 °C, Retention times were: 14.5 min (major), 16.6 min (minor). The submitters determined the enantiomeric excess of their product to be 86-88% using the following chiral HPLC conditions: Chiralcel OD, 250 × 4.6 mm (L × I.D.), solvent (isocratic) 85% hexanes, 15% isopropanol, flow rate 1.0 mL/min, wavelength λ = 220 nm. Retention times were: (1'
R,5'
S)-β-lactone, 18.06 min; (1'
S,5'
R)-β-lactone 22.96 min. An alternative method was developed previously
6a using chiral GC with a non-commercially available chiral column: 2,3-di-OAc-6-TBS-CD and showed 92% ee with
O-Ac quinidine as chiral promoter.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
This procedure describes an organocatalytic, enantioselective method for the asymmetric synthesis of bicyclic β-lactones. The intramolecular nucleophile-catalyzed aldol-lactonization (NCAL) process effectively merges catalytic, asymmetric carbocycle synthesis with β-lactone synthesis leading to unique bicyclic β-lactones.
6 The demand for concise synthetic routes to optically active β-lactones continues to grow due to continued development of novel transformations of these heterocycles and a reappraisal of their utility as synthetic intermediates
7 including natural product synthesis,
8 their continued occurrence in natural products,
9 their potent activity as enzyme inhibitors
10 and their utility as activity based probes.
11 β-Lactones can be viewed as "activated aldol products" since they possess the structural features of aldol products, yet they also have inherent reactivity due to ring strain (β-lactones, 22.8 kcal/mole; epoxides, 27.2 kcal/mole, Figure 1).
12
Figure 1. Comparison of structure and reactivity of β-lactones to aldol adducts and epoxides.
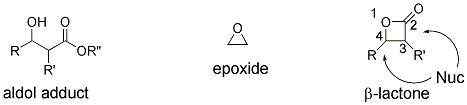
The aldol motif, the β-hydroxy carbonyl motif derived from polyketide biosynthesis, is common to many natural products; therefore asymmetric methods for their synthesis have been studied extensively. However, β-lactones arguably have greater utility over simple aldol products since they can undergo divergent nucleophilic cleavage processes occurring at either the acyl-oxygen (C
2-O
1) bond or the alkyl-oxygen (C
4-O
1) bond typically driven by release of ring strain. As a result, β-lactones undergo a number of interesting and useful stereospecific transformations making them versatile intermediates for organic synthesis.
7 Despite these facts, general methods for the direct synthesis of β-lactones in optically active form lag far behind those developed for epoxides and aldol adducts. Furthermore, there is much unexploited synthetic potential of β-lactones as chiral synthetic intermediates. Consequently, our group has been engaged in the development of efficient methods for the asymmetric synthesis of β-lactones and their subsequent transformations with applications to natural product synthesis.
13 Building on our initial reports of the NCAL process,
6we recently developed optimized conditions that renders the process more practical with the recognition of the importance of the counterion of the Mukaiyama reagent.
6c Several optically active cyclopentanes and cyclohexanes possessing fused β-lactones are now available in good yields and high enantioselectivities by this process (Table 1).
Table 1. Catalytic, Asymmetric Intramolecular NCAL Reactions Leading to Bicyclic β-Lactones
The utility of the dioxolane bicyclic β-lactone
5 described in this
Organic
Syntheses procedure was demonstrated in the synthesis of cyclopentane diol
13, a useful intermediate in the synthesis of antiviral carbocyclic nucleosides including (-)-aristeromycin (Scheme 1).
6a
Scheme 1. Utility of β-lactone 5 toward a formal synthesis of (-)-aristeromycin.
Highly diastereoselective NCAL reactions utilizing chiral aldehyde acids 15 and 18 also provide expedient access to functionalized bicyclic-β-lactones 16 and 19, respectively. Two examples are provided below, and the required aldehyde acid substrates are readily available from ozonolysis of (R)-citronellic acid (14, Scheme 2, eq 1) and from optically active β-hydroxy acid 18 readily obtained by Noyori reduction of ketoester 17 ultimately leading to the useful silyloxy substituted bicyclic-β-lactone 19 (Scheme 2, eq 2).
Scheme 2. β-Lactones via diastereoselective NCAL reactions of aldehyde acids.
The utility of the NCAL process was recently expanded to include the use of more tractable ketoacid substrates allowing access to bicyclic and tricyclic-β-lactones (Scheme 3).
15 This process was applied to an enantioselective synthesis of a reduced form of the DNA polymerase inhibitor, plakevulin A, and to a concise, 9-step bioinspired racemic and asymmetric syntheses of salinosporamide A and derivatives.
16 Furthermore, novel transformations
7 including dyotropic rearrangments
17 provide further avenues for exploitation of β-lactones in chemical synthesis.
Scheme 3. Extension of the NCAL process to ketoacid substrates leading to bicyclic and tricyclic-β-lactones with application to the synthesis of (+)-dihydroplakevullin.
A number of natural products possess bicyclic-β-lactones within their structures including salinosporamide A, omuralide (proteasome inhibitors),
b,c vibralactone (pancreatic lipase inhibitor), and spongiolactone (unknown activity).
18 In addition, cyclopentyl-fused, bicylic β-lactones can be envisioned as useful intermediates for the synthesis of several natural products including marine cembranoids such as verrillin and rameswaralide.
19
Figure 2. Some natural products possessing bicyclic β-lactones or structures potentially accessible from these intermediates.
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
(1,4-Dioxaspiro[4.5]dec-7-en-8-yloxy)trimethylsilane; (144810-01-5)
1,4-Cyclohexanedione mono-ethylene ketal: 1,4-Dioxaspiro[4.5]decan-8-one; (4746-97-8)
Trimethylsilyl chloride: Chlorotrimethylsilane; (75-777-4)
Triethylamine: Ethanamine, N,N-diethyl-; (121-44-8)
3-(2-(2-Oxoethyl)-1,3-dioxolan-2-yl) propanoic acid 360794-17-8
Triphenylphosphine; (603-35-0)
N-Methyl-2-chloropyridinium trifluoromethane sulfonate Pyridinium, 2-chloro-1-methyl-, 1,1,1-trifluoromethanesulfonate (1:1) 84030-18-2
2-Chloropyridine; (109-09-1)
Methyl trifluoromethanesulfonate; (333-27-7)
Quinidine: Cinchonan-9-ol, 6'-methoxy-; (56-54-2)
Diisopropylethylamine: 2-Propanamine, N-ethyl-N-(1-methylethyl)-; (7087-68
-5)
 |
Daniel Romo was born in San Antonio, Texas, USA in 1964. He received his B.A. in chemistry/biology from Texas A&M and a Ph.D. in Chemistry from Colorado State University as an NSF Minority Graduate Fellow under the tutelage of the late Professor Albert I. Meyers. Following postdoctoral studies at Harvard as an American Cancer Society Fellow, with Professor Stuart L. Schreiber he began his independent career at Texas A&M in 1993 and is currently Professor of Chemistry. In 2010 he became the Director of the Natural Products LINCHPIN Laboratory at Texas A&M University. Research interests in the Romo Group are at the interface of chemistry and biology focused on total synthesis and biomechanistic studies of natural products and the asymmetric synthesis and application of β-lactones in organic synthesis and their utility as both cellular probes and potential drug candidates. |
 |
Henry Nguyen was born in 1971 in Saigon, Vietnam. In 1993, he immigrated to America with his family and continued his undergraduate studies at Texas Woman's University. In 2000, he received his B.S. and then M.S. in chemistry in 2004 under the guidance of Professor James Johnson at Texas Woman's University. His research focused on mechanistic studies of methoxide ion substitution with Z and E isomers of methyl O-methylbenzothiohydroximates. In November 2004, he joined the research group of Prof. Romo at Texas A&M University and received his Ph.D. in 2010. His research focused on the enantioselective, biomimetic total synthesis and bioactivity of salinosporamide A and derivatives. |
 |
Seong Ho Ryan Oh was born in 1968 in Jeju, Korea and obtained his B.S. degree in 1991 and M.S. degree in 1993 under the direction of Kwan Soo Kim from Yonsei University in Seoul, Korea. In 1993, he joined the Chemical Research and Development group at LG Life Science, Taejon, Korea. In 2000, he continued as a Ph.D. student under the supervision of Prof. Romo at Texas A&M University. After completing his Ph.D. studies on the asymmetric synthesis of heterocycle-fused bicyclic β-lactones in 2005, he carried out postdoctoral research on the synthesis of orally active, antimalarial, anticancer, artemisinin-derived trioxane analogs in the laboratory of Gary Posner at Johns Hopkins University. In 2007, he joined the Biocatalyst and Chemical Development group at Codexis in Singapore. |
 |
Huda Henry-Riyad was born in 1974 in Khartoum, Sudan. In 2004 she obtained her Ph.D. from the University of Toronto under the supervision of Professor Thomas Tidwell. While at U of T, she examined ketenes and their reactivity with nitroxyl free radicals. In 2005 she joined the research group of Prof. Romo and worked on the synthesis of novel bicyclic β-Lactones via the nucleophile catalyzed aldol-lactonization (NCAL) process. She is currently the Director of The Key Research Center. The center's multi-disciplinary research idea is to bring different branches of knowledge together and to strengthen ties between social and the natural-physical sciences. Current research focuses on neuroscience and other related research areas that lead into the solution of mental, physical and social disorders. |
 |
Diana Sepulveda-Camarena was born in Guadalajara, Mexico. She attended the Instituto Tecnologico y de Estudios Superiores de Monterrey - Campus Monterrey where she received a B.Sc. in Chemistry. Prior to graduating in Fall 2006, she conducted her undergraduate research thesis project under the supervision of Prof. Romo at Texas A&M University. In February 2007, she joined BASF - Industrial Coatings division and worked in Apodaca, Mexico until she went back to Texas A&M in the Fall of 2008, where she is currently pursuing a Ph.D. in Chemistry under the supervision of Prof. Daniel Singleton. |
 |
Ms. Gerri E. Hutson obtained her B.S. in Chemistry in 2004 from Northern Illinois University, performing undergraduate research under Dr. Qingwei Yao. During her undergraduate studies she interned at Merck & Co. in Process Research in 2003 and 2004. She began her Ph.D. work under the supervision of Dr. Rawal in 2005, focusing her research on the utility of metal salens in enantioselective transformations. Her thesis title was "Modified Salen Catalyts in Atom-Economic Reactions: Enantioselective Carbonyl-ene and Nazarov Cyclization Reactions" and she was awarded a Ph.D. in Chemistry in 2010. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved