Prior and subsequent to our original report in
Organic Syntheses, we have described a number of highly diastereoselective Lewis acid-mediated additions of titanium enolates from chiral
N-acyl thiazolidinethiones to acetals.
2 These involve the addition of
N-propanoyl and
N-acetyl-4-isopropyl-1,3-thiazolidine-2-thione to dialkyl acetals (eq 1 and 2 in Scheme 1)
3-5 and dimethyl ketals from methyl ketones (eq 3 in Scheme 1).
6 Furthermore, the scope of the acyl groups has been recently expanded to
N-glycolyl derivatives. Especially,
O-pivaloyl protected
N-glycolyl 4-isopropyl-1,3-thiazolidine-2-thione undergoes highly diastereoselective additions to a wide array of dimethyl and dibenzyl acetals (eq 4 in Scheme 1).
7Scheme 1. Stereoselective additions of titanium enolates from N-acyl-4-isopropyl-1,3-thiazolidine-2-thione to dialkyl acetals and ketals
These reactions likely proceed through a mechanism in which an oxocarbenium intermediate approaches to the less hindered face of the titanium enolate. Taking advantage of such a picture, this chemistry has been successfully applied to glycals. Thereby, activation of glycals by Lewis acids triggers the formation of cyclic and conjugated oxocarbenium intermediates that may participate in highly diastereoselective α- and β-
C-glycosidation processes with the abovementioned titanium enolates. Importantly, the appropriate choice of the chiral auxiliary and the C6-protecting group determines the stereochemical outcome of these carbon-carbon bond forming reactions and permits the modular preparation of three of the four possible diastereomers of α- or β-1'-methyl-substituted
C-glycosides (Scheme 2).
8-10Scheme 2. Stereoselective C-glycosidation reactions
Synthetic applications
These methods have been already applied to the synthesis of natural products. For instance, our group took advantage of the Lewis acid-mediated addition of the titanium enolates from (
S)
N-acetyl and
N-propanoyl 4-isopropyl-1,3-thiazolidine-2-thiones (
1 and
2 respectively in Scheme 3) to dialkyl acetals for the construction of the C9-C21 fragment of debromoaplysiatoxin.
11 As shown in Scheme 3, chromatographic purification of the product formed from the reaction between
1 and the dimethyl acetal of an aromatic aldehyde afforded diastereomerically pure adduct
3 in 82% yield. Removal of the chiral auxiliary gave alcohol
4, which was further elaborated to chiral dibenzyl acetal
5. Then, the stage was set for the introduction of two new stereocenters. Indeed, the asymmetric induction imparted by the titanium enolate of
2 and the
Felkin bias of the oxocarbenium cation from
5 provided adduct
6 as a single diastereomer (dr > 98:2) in 74% yield. Finally, treatment of
6 with MeNHOMe furnished Weinreb amide
7 in 90% yield.
12Scheme 3. Stereoselective synthesis of C9-C21 fragment of debromoaplysiatoxin
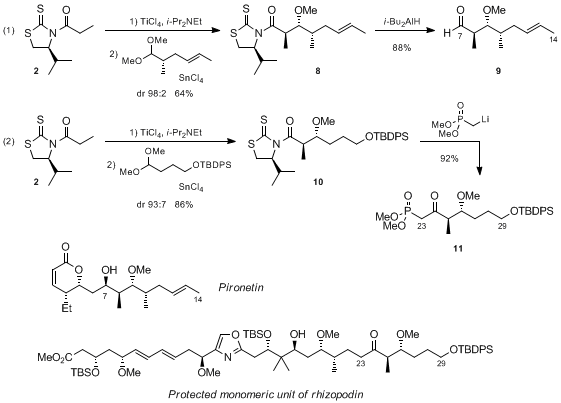
Crimmins and Chakraborty have also used these transformations in the total syntheses of pironetin and a protected form of the monomeric unit of rhizopodin respectively (Scheme 4).
13,14 Thereby, Crimmins reported that the addition of
2 to a chiral dimethyl acetal provided adduct
8 as a single diastereomer in 64% yield, which was further converted into aldehyde
9 by treatment with
i-Bu
2AlH (eq 1 in Scheme 4).
13 In turn, Chakraborty described a similar process involving an achiral dimethyl acetal in which adduct
10 was obtained in an excellent diastereomeric ratio (dr 93:7) and 86% yield (eq 2 in Scheme 4).
14 Interestingly, removal of the chiral auxiliary by the lithium salt of the dimethyl methylphosphonate furnished ketophosphonate
11 ready to participate in an HWE olefination.
Scheme 4. Total syntheses of pironetin and a monomeric unit of rhizopodin
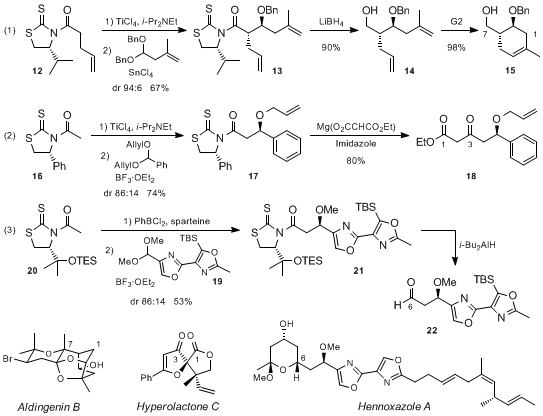
Other reports have also established that these Lewis acid acid-mediated couplings of
N-acyl thiazolidinethiones and acetals are flexible enough to be successfully adapted to the synthesis of a broad array of natural products. For instance, Crimmins took advantage of the highly diastereoselective reaction of the titanium enolate from
N-(4-pentenoyl) thiazolidinethione
12 and a dibenzyl acetal, the mild removal of the chiral auxiliary from adduct
13, and the RCM of the resultant alcohol
14 to obtain in a few steps the cyclic core
15 of aldigenin B (eq 1 in Scheme 5).
15 In turn, Hodgson reported that the addition of the titanium enolate of (
R)-phenylglycine-derived
N-acetyl thiazolidinethione
16 to the benzaldehyde diallyl acetal provided diastereomerically pure adduct
17 in 74% yield after chromatography. Further treatment of this adduct with magnesium acetoacetate and imidazole led to β-keto ester
18, a key intermediate in the total synthesis of hyperolactone C (eq 2 in Scheme 5).
16 Finally, the synthesis of hennoxazole A by Smith includes a comprehensive study on the Lewis acid-mediated additions of
N-acetyl thiazolidinethiones to the dimethyl acetal of bisoxazole
19.
17 This revealed that the ability of an oxazole nitrogen atom to coordinate with the titanium center altered the stereochemical outcome of such additions and the undesired diastereomer was obtained in 80:20 diastereomeric ratio and 67% yield. Looking for alternative conditions, Smith found that the enolization of
N-acetyl thiazolidinethione
20 using Sammakia's conditions (PhBCl
2/sparteine) followed by the BF
3·OEt
2-mediated addition of the resultant boron enolate to
19 provided the desired adduct
21 in 86:14 diastereomeric ratio and 53% yield (eq 3 in Scheme 5). Eventually, reduction of
21 with
i-Bu
2AlH gave aldehyde
22, which was immediately used in the next step.
Scheme 5. Total syntheses of aldigenin B, hyperolactone C, and hennoxazole
Dialkyl acetals are not the only suitable substrates for these transformations. Certainly, oxocarbenium cations from cyclic hemiacetals and glycals (see Scheme 2) can also undergo highly diastereoselective additions to titanium enolates from
N-acyl thiazolidinethiones, as was used by our group in the stereoselective synthesis of the western hemisphere of salinomycin. Indeed, the SnCl
4-mediated addition of the titanium enolate from
N-butanoyl thiazolidinethione
23 to hemiacetal
24 afforded α-
C-glycoside
25 as a single diastereomer (dr > 97:3), which was treated with methanol to obtain methyl ester
26 in 75% overall yield (eq 1 in Scheme 6).
18 More recently, Leighton has reported that the coupling of structurally complex
O-acetyl hemiacetal
27 with the titanium enolate of
ent-2 produced α-
C-glycoside
28 as a single diastereomer in an outstanding 91% yield (eq 2 in Scheme 6).
19 Methanolysis proceeded exceptionally smoothly to give
29 in quantitative yield, which was finally converted into zincophorin methyl ester in a few steps.
Scheme 6. Syntheses of antibiotic polyethers
In summary, the Lewis acid-mediated addition of titanium enolates from N-acyl thiazolidinethiones to dialkyl acetals, hemiacetals, or glycals represents a powerful synthetic tool for the stereoselective construction of carbon-carbon bonds leading to anti α-alkyl-β-alkoxy carboxylic derivatives. Importantly, the chiral auxiliary can be easily removed using a number of mild conditions, which confers to these methodologies a remarkable appeal for the synthesis of natural products.
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved