Org. Synth. 2013, 90, 200-214
DOI: 10.15227/orgsyn.090.0200
Nickel-Catalyzed Cross-Coupling of Aryl Halides with Alkyl Halides: Ethyl 4-(4-(4-methylphenylsulfonamido)-phenyl)butanoate
Submitted by Daniel A.Everson, David T.George, and Daniel J.Weix*
1.
Checked by Jonas F.Buergler and John L.Wood.
1. Procedure
A. N-(4-Bromophenyl)-4-methylbenzenesulfonamide (1) (Note 1). With no precautions to exclude air or moisture, a 500-mL round-bottomed flask is equipped with a PTFE-coated egg-shaped magnetic stir bar (38 x 16 mm). 4-Bromoaniline (Note 2) (15.1 g, 88.0 mmol, 1.00 equiv) and p-toluenesulfonyl chloride (Note 3) (17.1 g, 89.8 mmol, 1.02 equiv) are weighed into plastic weigh boats and added to the vessel through a powder funnel. Dichloromethane (Note 4) (230 mL) is then poured into the vessel from a graduated cylinder, followed by the addition of pyridine (Note 5) (7.8 mL, 97 mmol, 1.1 equiv) by syringe in two portions. The reaction vessel is stoppered with a rubber septum and an 18-gauge needle to vent the reaction to air before stirring (~500 rpm) at room temperature (20 °C) for 16 h.The reaction mixture turns from yellow to clear orange immediately after the addition of pyridine.After the reaction is judged complete by TLC analysis (Note 6), the reaction mixture is poured into a 2 L separatory funnel containing 800 mL of sat. NH4Cl(aq) (Note 7). The organic layer is separated and the aqueous extracted with dichloromethane (2 x 50 mL). The combined organic layer is then washed with water (400 mL), brine (400 mL) and dried over anhydrous MgSO4 (10.0 g, ~1 min) (Note 8).The drying agent is removed by vacuum filtration through a course-fritted glass Büchner funnel.The filtrate is concentrated by rotary evaporation (30 °C, 74 mmHg) to yield 28.5-28.6 g (99% yield) of N-(4-bromophenyl)-4-methylbenzenesulfonamide as a white solid (Note 9).
B. Ethyl 4-(4-(4-methylphenylsulfonamido)phenyl)butanoate (2). A 500-mL 3-necked Morton flask with 24/40 ground-glass joints is equipped with a nitrogen-gas inlet on the left neck, a glass stopper on the right neck, and the center neck is equipped for mechanical stirring (glass bearing, glass stirrer rod, 60 mm PTFE paddle, RW 20 Tekmar motor) (Note 10). NiI2.xH2O (528 mg, 1.31 mmol, 0.051 equiv) (Note 11), 4,4'-di-methoxy-2,2'-bipyridine (284 mg, 1.31 mmol, 0.051 equiv) (Note 12), sodium iodide (1.25 g, 8.34 mmol, 0.33 equiv) (Note 13), and N-(4-bromophenyl)-4-methylbenzenesulfonamide (8.33 g, 25.5 mmol, 1.00 equiv) are transferred via powder funnel through the right neck of the Morton flask.To these solids, 1,3-dimethylpropyleneurea (DMPU, 105 mL) (Note 14) is added from a graduated cylinder.Then pyridine (105 μL, 1.31 mmol, 0.053 equiv) (Note 5) and ethyl 4-bromobutyrate (4.0 mL, 28 mmol, 1.1 equiv) (Note 15) are added by syringe.A gas outlet adapter with tubing attached to an oil-filled bubbler is placed in the right neck before submerging the reaction vessel up to the solvent line in an oil bath pre-equilibrated to 60 °C.The vessel headspace is purged with nitrogen for 15 min while the mixture is stirred (500-600 rpm) (Note 16).After the nitrogen purge is complete, the gas outlet on the right neck is replaced with the original glass stopper and the reaction mixture is stirred at 60 °C until the reaction mixture takes on a dark green color (15-30 min) (Note 17). Next, zinc powder (6-9 μm, 3.44 g, 52.6 mmol, 2.06 equiv) (Note 18) is added through a powder funnel attached to the right neck under a positive flow of nitrogen.The flask is kept under a slight positive pressure of nitrogen during the course of the reaction.Generally within 5-15 min after the addition of zinc, the reaction undergoes a characteristic dark green to orange-brown color change, indicating the reaction has begun.The reaction is judged complete when the color changes to black (Note 19).
Once the reaction mixture changes color to black, it is allowed to cool to room temperature before filtering though a pad of diatomaceous earth (15 g, wetted with 40 mL of diethyl ether and packed firmly in place) (Note 20) in a 350-mL course-fritted glass Büchner funnel.The reaction vessel is rinsed with ether (3 × 50 mL) (Note 21) and these rinses are filtered as well.The filtrate is poured into a 1 L separatory funnel containing 150 mL of 1 M NH4Cl(aq). The organic layer is separated and set aside.The aqueous layer is then extracted with additional ether (3 × 60 mL). The combined organic layers are washed with water (75 mL) and brine (75 mL), then dried over MgSO4 (15 g, ~1 min). The solids are removed by vacuum filtration through a course-fritted glass Buchner funnel and washed with ether (20 mL). The filtrate is then concentrated by rotary evaporation (30 °C, 74 mmHg) to give a colorless oil.The aqueous layers are combined and set aside for later recovery of DMPU, if desired (Note 22).
The oil is purified by stepped-gradient flash chromatography on silica gel (column diameter 90 mm, 225 g SiO2) (Note 23).The column is dry-packed with silica gel, equilibrated with hexanes (500 mL, passed through the column twice) (Note 24), and the compound is loaded directly onto the silica with dichloromethane (3 mL). The vessel containing the crude material is rinsed with 95:5 hexanes:EtOAc (5 mL), and additional dichloromethane (5 mL), which are then used to rinse the sides of the column (Note 25). After adding a layer of sand (~12 mm.) to the column, the compound is eluted with the following ratios (v:v) of hexanes:EtOAc: 95:5 (500 mL, discarded to forerun), 90:10 (500 mL, discarded to forerun), 85:15 (500 mL, discarded to forerun), 80:20 (500 mL), 75:25 (1000 mL), 70:30 (500 mL), 65:35 (500 mL), and 60:40 (500 mL) (Note 26).Fractions are collected in 25 mm x 150 mm test-tubes (~65-70 mL).The fractions containing only product as determined by TLC (Note 19) are rinsed with EtOAc into a tared 1 L recovery flask and concentrated by rotary evaporation (35 °C, 74 mmHg).A small, tared PTFE-coated magnetic stir bar (7 mm x 2 mm) is added to the viscous, colorless oil before drying on high vacuum (0.1 mmHg, 20 °C) for 1 h to remove any traces of solvent.A heat gun is used to gently warm the oil while on high vacuum and the heat is removed as soon as gas bubbles begin to evolve.After drying, the flask contained 7.16-7.70 g (78%-83% yield) (Note 27) of ethyl 4-(4-(4-methylphenylsulfonamido)phenyl)-butanoate as a colorless viscous oil.
2. Notes
1.
This procedure is a modification of a literature method.
22.
4-Bromoaniline (98+%, Alfa Aesar or 97%, Sigma-Aldrich) was used a received.
3.
p-Toluenesulfonyl chloride (98%, Alfa Aesar) was used as received.The submitters used 1.10 equivalents of
p-toluenesulfonyl chloride, but in the hands of the checkers this lead to contaminated product; therefore, the checkers decreased the amount to 1.02 equivalents.This discrepancy may be due to the use of
p-toluenesulfonyl chloride of different quality since the submitters used different vendors (98%, Lancaster or ≥98% Sigma-Aldrich).
4.
Dichloromethane (certified ACS grade, Fischer Scientific) was used as received.
5.
Pyridine (ultrapure, spectrophotometric grade 99.5+%, Alfa Aesar) was used as received.
6.
The progress of the reaction can be followed by TLC analysis (eluent: 4:1 hexanes:EtOAc, visualization with 254 nm UV light).
p-Toluenesulfonyl chloride has an R
f of 0.58, 4-bromoaniline has an R
f of 0.23, and
N-(4-bromophenyl)-4-methylbenzenesulfonamide (
1) has an R
f of 0.28.TLC analysis was performed on pre-coated glass plates (SiliaPlate, 60Å, 250 μm, F
254, SiliCycle).The submitters used EMD silica gel 60 F
254 pre-coated glass plates.
7.
Ammonium chloride (ACS certified, crystalline, Malinckrodt) was used as received.
8.
Magnesium sulfate (anhydrous powder, Fisher Scientific) was used as received.
9.
In the hands of the checkers the yield was greater than 99% and the product obtained as a white solid.The submitters report that in their case the yield of this procedure was consistently greater than 80%, but the color of the final product would vary from faint yellow to brown.The color of the material did not affect the outcome of the next reaction.The product exhibited the following physiochemical properties and was stable on the bench top for months: mp: 148-149 °C;
1H NMR
pdf (400 MHz; CDCl
3) δ: 2.36 (s, 3 H), 6.96 (d,
J = 8.6 Hz, 2 H), 7.22 (d,
J = 7.8 Hz, 2 H), 7.31 (s, 1 H), 7.31 (d,
J = 9.0 Hz, 2 H), 7.66 (d,
J = 8.6 Hz, 2 H).
13C NMR
pdf (101 MHz; CDCl
3) δ: 21.6, 118.4, 122.9, 127.2, 129.8, 132.3, 135.5, 135.7, 144.2. FTIR (film, cm
-1): 3254 (N-H), 1328 and 1162 (S=O).HRMS (
m/
z) (ESI) calc.for C
13H
13BrNO
2S (M+H
+) 327.9825, found 327.9823.Anal.Calcd.for C
13H
12BrNO
2S: C, 47.86; H, 3.71; N, 4.29; found: C, 47.96; H, 3.83; N, 4.40.
10.
The stirrer paddle must not be in contact with the bottom or sides of the flask.The stirrer paddle was generally held ~10 mm above the bottom of the flask to prevent mechanical activation of the reducing agent, which results in greater amounts of hydrodehalogenation by-products and lower product yields. PTFE sleeves (ribbed, 24/40, Fischer Scientific) were used to seal all ground glass joints, and heavy mineral oil (mineral oil white, heavy, Malinckrodt) was used to lubricate the glass stir rod inside the 24/40 standard taper adapter (see photo).The checkers used thin PTFE sleeves.
11.
Nickel (II) iodide hydrate was purchased from Strem Chemicals and used as received.The submitters used elemental analysis to assess the water content to be
x = 3.5, but, because a slight excess of nickel iodide does not change the outcome of these reactions, the molecular weight of the nickel (II) iodide was taken to be that of the pentahydrate (MW = 402.58).For further information, see discussion section.
12.
4,4'-Di-methoxy-2,2'-bipyridine (97%, Sigma-Aldrich) was used as received.
13.
Sodium iodide (puriss.p.a., ≥99%, Sigma-Aldrich) was used as received.The submitters used Strem Chemicals (anhydrous, 99%).
14.
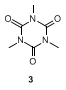
The submitters state that 1,3-dimethylpropylene urea (DMPU) (99%, AK Scientific, Inc.) was used as received.The DMPU can be recovered and reused for subsequent cross-coupling reactions (see Note
22).In the case of the checkers, the use of undistilled DMPU (99%, AK Scientific, Inc.or 98%, Alfa Aesar) resulted in significant amounts of a side product.According to NMR and mass analyses this contamination has been tentatively identified as 1,3,5- trimethyl-1,3,5-triazine-2,4,6-(1H,3H,5H)trione (
3) and is clearly arising from the DMPU, since it can be detected by simply extracting DMPU with water and diethyl ether.The fact that this contamination has the same R
f as
2 makes it difficult to remove by column chromatography.Distillation of the commercially available DMPU as described by the submitters
(Note 22) decreased the amount of this side product significantly; however, trace amounts (0.5 mol % for the submitters) were always visible in the NMR spectrum (s, 3.35 ppm).
15.
Ethyl 4-bromobutyrate (98%, Alfa Aesar) was used as received.
16.
The submitters used argon instead of nitrogen and a rubber septum pierced with a needle attached to the oil bubbler to purge the vessel.Oxygen does not irreversibly decompose the catalyst because the stoichiometric reducing agent can reduce any oxidized catalyst.However, reactions with too much oxygen in the headspace often suffer from long induction periods that prevent the catalyst from reacting with starting materials until the oxygen in the headspace has finished reacting with the catalyst and reducing agent.Sometimes reactions do not proceed to completion in a reasonable amount of time (< 24 h) when there is a large headspace volume of air.See discussion section below.
17.
In the hands of the submitters the color change to deep green was finished in about 15 minutes.The pre-stirring allows the nickel iodide to coordinate to the bipyridine complex and form the dark-green, tetrahedral (4,4'-dimethoxy-2,2'-bipyridine)NiI
2.
18.
Zinc dust (<10 μm, ≥98%, Sigma-Aldrich) was used as received.The submitters used zinc powder (6-9 μm, 97.5% metal basis, Alfa Aesar).
19.
The submitters have found the color change to be a reliable method for determining the end-point of the reaction.For this particular substrate the red-orange to black color change occurred in 4-6 h.No further product formation was observed if the reaction was allowed to stir longer (up to 48 h), and no decomposition of the product was observed either.In the hands of the checkers the color change occurred after 3-5 h.The progress of the reaction can be followed by TLC analysis (eluent: 4:1 hexanes:EtOAc, visualization with 254 nm UV light).
N-(4-Bromophenyl)-4-methylbenzenesulfonamide (
1) has an R
f of 0.23, ethyl 4-(4-(4-methylphenylsulfonamido)phenyl)butanoate (
2)
has an R
f of 0.15 and 4-methyl-
N-phenylbenzenesulfonamide (hydrodehalogenated aryl bromide) has an R
f of 0.25.Since the starting material
1 and the hydrodehalogenated side product have very similar R
f judging the progress of the reaction by TLC is difficult; therefore, monitoring the color change is a good alternative method.
20.
Filter agent, Celite® 545 was purchased from Sigma-Aldrich and used as received.The submitters used "Celite 545 filter aid" from Fischer Scientific.
21.
Diethyl ether (certified ACS grade, 7 ppm BHT stabilizer, Fisher Scientific) was used as received.
22.
To the combined aqueous layers from the workup, sodium chloride (Aldrich) was added until the solution was saturated (generally ~ 5 g, some undissolved salt does not pose a problem).DMPU was extracted from the salted aqueous layer with dichloromethane (3 × 75 mL).The combined organic layers were dried over magnesium sulfate (15 g, ~1 min), and the magnesium sulfate was removed by vacuum filtration through a course-fritted glass Büchner funnel.The filtrate was concentrated by rotary evaporation (35 °C, 73 mmHg) to give 90-95 mL (86%-90% recovery) of faintly yellow liquid.This material was then distilled from calcium hydride (Sigma-Aldrich, reagent grade, 95%) (60-61 °C, 0.05 mmHg)
3, dried to <1000 ppm water over 4 Å molecular sieves, and reused in subsequent cross coupling reactions.The submitters repeated the title cross coupling reaction on identical scale and obtained 8.63 g (94%) of ethyl 4-(4-(4-methylphenylsulfonamido)phenyl)butanoate (
2) using DMPU recycled by this method.
23.
Silica gel (SiliaFlash® P60, 230-400 mesh, SiliCycle) was used as received.The submitters used EMD silica gel 60 (mesh 230-400)
24.
Hexanes (certified ACS grade, 4.2% various methylpentanes, Fisher Scientific) were used as received.
25.
Ethyl acetate (EtOAc, certified ACS grade) was purchased from Fischer Scientific and used as received.
26.
The separation between product and hydrodehalogenated aryl bromide is small (ΔR
f = 0.1).The submitters point out, therefore, that the yield may vary 5-15% depending on the number of mixed fractions.The checkers obtained only few mixed fractions, not calculating the possible yield loss.
27.
The submitters obtained the product as faintly yellow oil in 79-93% yield.The submitters also mention that the product oil will slowly solidify after scratching the sidewall of the vessel with a metal spatula.Both the oil and the resulting white solid had identical spectral properties.In the hands of the checkers the product solidified only in one case after a prolonged time in the freezer.The product exhibited the following physiochemical properties and was stable on the bench top for months:
1H NMR
pdf (400 MHz; CDCl
3) δ: 1.22 (t,
J = 7.2 Hz, 3 H), 1.85 (quintet,
J = 7.5 Hz, 2 H), 2.24 (t,
J = 7.4 Hz, 2 H), 2.34 (s, 3 H), 2.54 (t,
J = 8.0 Hz, 2 H), 4.09 (q,
J = 7.0 Hz, 2 H), 6.96-7.02 (m, 4 H), 7.05 (s, 1 H), 7.19 (d,
J = 8.6 Hz, 2 H), 7.63 (d,
J = 8.2 Hz, 2 H).
13C NMR
pdf (101 MHz; CDCl
3) δ: 14.2, 21.5, 26.4, 33.5, 34.4, 60.3, 122.0, 127.2, 129.2, 129.6, 134.4, 136.1, 138.6, 143.7, 173.5. FTIR (film, cm
-1) 3255 (N-H); 1731 (C=O); 1337; 1162 (S=O).HRMS (
m/
z) (ESI) calc.for C
19H
27N
2O
4S (M+NH
4+) 379.1687, found 379.1687.Anal.Calcd.for C
19H
23NO
4S: C, 63.13; H, 6.41; N, 3.88; found (oil): C, 62.97; H, 6.54; N, 3.98.mp (of crystallized material obtained by the submitters): 49-52 °C.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Very recently, our group reported a new nickel-catalyzed method to directly couple haloalkanes (iodides or bromides) with haloarenes (iodides, bromides, or electron-poor chlorides).
4-5 A distinguishing feature of this method is that the only organometallic intermediates are catalytic organonickel species – no organozinc or organomanganese reagents are involved.Previous transition metal-catalyzed methods to synthesize alkylated aromatic compounds have relied on the coupling of pre-formed aryl nucleophiles with alkyl electrophiles,
6-8 aryl electrophiles with pre-formed alkyl nucleophiles,
9 or
in situ formation of a carbon nucleophile.
10-15 A major advantage of this new approach is that most carbon nucleophiles are synthesized from the corresponding organic halides,
16 and thus a synthetic step can be eliminated.Additionally, the absence of stoichiometric strong nucleophiles or bases to aid trans-metalation imparts excellent functional group compatibility.Concurrent with our work, Gosmini and Amatore developed a similar cobalt-catalyzed method, though the mechanism remains unclear.
17 Following our studies, Peng has studied the use of a simplified catalyst for inter- and intramolecular couplings under similar conditions
18 and Gong noted that the addition of MgCl
2 can improve yields with secondary alkyl halides.
19Acidic groups, such as
N-arylsulfonamides (pKa similar to acetic acid in DMSO),
20 are well tolerated (title compound
2).Substrates that are prone to β-elimination when metalated (
6) couple cleanly with aryl bromides containing acidic protons (
5) (Table 1, entry 1) in stark contrast to the difficulties conventional cross-coupling methods have with these types of substrates.
21 Carbon nucleophiles used for Suzuki,
22 Stille,
23 and Hiyama-Denmark
24 cross-coupling reactions are not reactive under these conditions, allowing for the straightforward synthesis of poly-substituted aromatic compounds (Table 1, entries 4-6).Electrophilic groups, such as alkyl-aryl ketones, trifluoromethanesulfonic acid esters, and acetylated phenols, are compatible with our method as well (Table 1, entries 7-9).Lastly, the coupling of
26 with
27 is the synthetic equivalent of the α-arylation of acetaldehyde (Table 1, entry 10).
Scaling our recent procedures from 1 mmol to 25 mmol required addressing two difficulties, the heterogeneous nature of the reduction and variable induction periods that resulted in unpredictable reaction times.
The best results are obtained with mechanical stirring, but care must be taken to avoid grinding the zinc dust with the stirrer.Mechanical activation of metal powders is well known
25-27 and in the present reaction results in hydrodehalogenation products.This is presumably due to direct insertion of the zinc into the starting materials.Keeping the stirrer above the bottom surface and using a Morton flask
28 to ensure turbulent mixing results in reliable, high yields on large scale.Zinc powder from Alfa Aesar has proven more reliable than from other suppliers.If very deactivated zinc must be used, then brief activation with hydrochloric acid can be effective.
5As we discovered when conducting kinetic studies on the present reaction,
5 oxygen in the headspace leads to long induction periods.Although the reactions proceed as expected after this induction period on small scale, in some large scale reactions we observed that the starting materials would decompose or the catalyst would become inactivated before reactions could complete.Sweeping most of the oxygen out with argon (or nitrogen) enables consistent reaction times and higher yields.These reactions are oxygen
tolerant in that trace oxygen is not a concern, but large amounts of oxygen should be avoided.
Finally, although DMPU is routinely used on scale, its price is higher than other dipolar aprotic solvents routinely used in the lab.In order to conserve resources in our own lab, we worked out a simple procedure for recovering the DMPU that allows for efficient recycling of the solvent.
Table 1. Scope of Nucleophile Free Cross Couplinga
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
4-Bromoaniline; (106-40-1)
p-Toluenesulfonyl chloride: 4-Methylbenzene-1-sulfonyl chloride; (98-59-9)
Pyridine; (110-86-1)
NiI2.xH2O: Nickel(II) iodide hydrate; (7790-34-3)
4,4'-Di-methoxy-2,2'-bipyridine: 4,4'-di-methoxy-2,2'-bipyridine; (17217-57-1)
Sodium iodide; (7681-82-5)
1,3-Dimethyl-propylene urea: 1,3-Dimethyltetrahydropyrimidin-2(1H)-one; (7226-23-5)
Ethyl 4-bromobutanoate; (2969-81-5)
Zinc powder: Zinc; (7440-66-6)
 |
Daniel J.Weix was born and raised outside of Milwaukee, Wisconsin.He studied chemistry at Columbia University (BA, 2000), where he worked on helicene chemistry with Prof.Tom Katz.Graduate studies at the University of California, Berkeley (2000-2005) with Prof.Jon Ellman focused on the synthesis and application of tert-butanesulfinamide.After postdoctoral studies (2005-2008) on Ir-catalyzed allylation chemistry with Prof.John Hartwig at both Yale University and the University of Illinois, Daniel started his independent career at the University of Rochester in 2008.His research program focuses on the development of new concepts in catalysis, with a major current interest in cross-electrophile coupling reactions. |
 |
Daniel A.Everson was born in Minneapolis, Minnesota in 1985.He completed his B.S.in 2007 at the University of St.Thomas, St.Paul, Minnesota, working with Prof.J.Thomas Ippoliti.After working as a research associate for Prof.Ippoliti during 2007-2008 he joined the group of Daniel J.Weix at the University of Rochester, Rochester, New York in 2008, and was awarded an NSF graduate research fellowship in 2010.His research interests are in developing new transition metal mediated nucleophile free cross coupling methodologies. |
 |
David T.George was born in Highland Park, Illinois in 1990 and grew up in Middletown, New Jersey.He will complete his B.S.at the University of Rochester, Rochester, New York, in May 2013 and joined the group of Daniel J.Weix in 2011.In addition to research at the University of Rochester, David also worked for Burpee Materials Technology, Eatontown, New Jersey.His current research project is the application of reductive coupling reactions to natural product synthesis. |
 |
Jonas F.Buergler was born in Davos, Switzerland in 1982.He obtained his BSc and MSc in Chemistry (2006) from the Swiss Federal Institute of Technoloy (ETH) Zurich.In 2011 he was awarded his PhD from the ETH for his work on the synthesis and applications of P-stereogenic ferrocenyl phosphines under the supervision of Prof- Dr.A.Togni.Since September 2011 he is a postdoctoral fellow in the group of Prof.John L.Wood with fellowships from the Swiss National Science Foundation and the Novartis Jubilee Foundation, where he is currently working on the total synthesis of tetrapetalone A.
|
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved