Org. Synth. 2013, 90, 271-286
DOI: 10.15227/orgsyn.090.0271
Allyl Cyanate-To-Isocyanate Rearrangement: Preparation of tert-Butyl 3,7-dimethylocta-1,6-dien-3-ylcarbamate
Submitted by Yoshiyasu Ichikawa,
1 Noriko Kariya, and Tomoyuki Hasegawa.
Checked by Eiji Yoshida and Tohru Fukuyama.
1. Procedure
A. (E)-3,7-Dimethylocta-2,6-dien-1-ylcarbamate (3): An oven-dried 500-mL, three-necked, round-bottomed flask (Note 1) equipped with a magnetic stir bar (8 x 30 mm), a thermometer with a Teflon holder, a rubber septum and an inlet adapter with a three-way stopcock fitted with an argon inlet is charged with geraniol (1) (11.0 g, 71.3 mmol, 1.0 equiv) (Note 2) and dry dichloromethane (180 mL) (Note 3). The flask is immersed in a sodium chloride/ice bath (internal temperature -15 °C). Trichloroacetyl isocyanate (9.30 mL, 14.7 g, 78.0 mmol, 1.10 equiv) (Note 4) is added slowly via a syringe through the rubber septum over a 10 min period at a rate to maintain the internal temperature below -10 °C. After stirring at -15 °C (internal temperature) for 20 min (Note 5), methanol (0.90 mL, 0.71 g, 22 mmol, 0.30 equiv) (Note 6) is added via a syringe inserted through the rubber septum to quench excess trichloroacetyl isocyanate (Note 7). The solution is transferred to a 1-L recovery flask with the aid of dichloromethane (15 mL), and then concentrated by rotary evaporation (200 mmHg, water bath temperature 40 °C) and dried under vacuum (1.0 mmHg) (Note 8) to yield trichloroacetyl carbamate 2 as a pale yellow oil (25.9 g). The crude 2 in the 1-L recovery flask is dissolved in methanol (240 mL) (Note 6) and a magnetic stir bar (8 x 30 mm) is placed in the flask. Aqueous K2CO3 (2 M, 140 mL) is added to the stirred mixture in portions at room temperature over a 10 min period, resulting in formation of a cloudy solution (Note 9). After stirring at room temperature for 3 h while checking the consumption of trichloroacetyl carbamate 2 by TLC analysis (Note 5), the reaction mixture is concentrated by rotary evaporation (70 mmHg, water bath temperature 40 °C) to about half the original volume. The resulting cloudy solution is transferred to a 1-L separatory funnel and extracted with diethyl ether (3 x 50 mL) (Note 10). The combined organic extracts are washed with water (50 mL) and brine (50 mL), dried over anhydrous MgSO4 and filtered through cotton. After removal of the solvent by rotary evaporation (70 mmHg, water bath temperature 40 °C), the oily residue is dried under vacuum (1.0 mmHg) (Note 8) to constant weight, affording crude carbamate 3 (13.93 g, 99%) (Note 11) as a white solid, which is used in Step B without further purification.
B. t-Butyl 3,7-dimethylocta-1,6-dien-3-ylcarbamate (6). The crude carbamate 3 (13.93 g, 70.6 mmol, 1.0 equiv) in a 300-mL recovery flask is dissolved in toluene (10 mL) (Note 12) and the solvent is removed azeotropically by rotary evaporation (70 mmHg, 50 °C). After repeating this procedure once more followed by drying under vacuum (1.0 mmHg) (Note 8) for 30 min, crude 3 is dissolved in dry dichloromethane (2 x 10 mL) and transferred by a pipette to a 500-mL three-necked, round-bottomed flask equipped with a stir bar (8 x 30 mm), a thermometer with a Teflon holder, a 50-mL pressure-equalizing addition funnel with a rubber septum, and an inlet adapter with a three-way stopcock fitted with an argon inlet. The flask is charged with dry dichloromethane (180 mL), triphenylphosphine (28.0 g, 107 mmol, 1.5 equiv) (Note 13) and Triethylamine (20.0 mL, 14.5 g, 143 mmol, 2.0 equiv) (Note 14). The flask is cooled with a sodium chloride/ice bath (internal temperature -15 °C). Carbon tetrabromide (37.8 g, 114 mmol, 1.6 equiv) (Note 15) in dry dichloromethane (20 mL) is added via the dropping funnel over a 30 min period, maintaining the internal temperature below -10 °C. The dropping funnel is rinsed with dry dichloromethane (5 mL). After stirring at -15 to -10 °C (internal temperature) for 20 min (Note 16), the cooling bath is removed and the yellow reaction mixture is diluted with n-hexane (150 mL) (Note 17). After stirring at room temperature for 30 min, the light-brown solution is poured into aqueous KHSO4 (1 M, 300 mL) in a 1-L Erlenmeyer flask equipped with a stir bar (8 x 30 mm). The three-necked flask is rinsed with n-hexane (2 x 10 mL). After stirring vigorously for 30 min, the solution is transferred into a 1-L separatory funnel. The Erlenmeyer flask is rinsed with n-hexane (2 x 10 mL). The organic layer is separated, washed with water (200 mL), saturated aqueous NaHCO3 (100 mL) and brine (200 mL), dried over anhydrous MgSO4 and filtered through cotton. Concentration by rotary evaporation (200 mmHg, 40 °C) gives a crude mixture containing allyl isocyanate 5 and triphenylphosphine oxide as a yellow solid (72.8 g), to which is added n-hexane (200 mL). The resulting mixture is swirled in a sonicator for 5 min to form a suspension. After filtration through a pad of Celite (Note 18) and washing the filter cake with n-hexane (3 x 50 mL), the combined golden yellow filtrate is concentrated by rotary evaporation (90 mmHg, water bath temperature 40 °C), and then dried under vacuum (1.5 mmHg) for 30 min to provide crude isocyanate 5 (25.4 g) as a pale yellow oil (Note 19 and Note 20).
An oven-dried 1-L, three-necked, round-bottomed flask equipped with a stir bar (8 x 30 mm), a thermometer with a Teflon holder, a 100-mL dropping funnel with a rubber septum, and an inlet adapter with a three-way stopcock fitted with an argon inlet is charged with dry tetrahydrofuran (280 mL) (Note 21) and tert-butyl alcohol (68.0 mL, 52.3 g, 711 mmol, 10.0 equiv) (Note 22). After cooling to 0 °C in an ice bath, lithium hexamethyldisilazide (1 M solution in tetrahydrofuran, 71.3 mL, 71.3 mmol, 1.0 equiv) (Notes 23 and 24) is added via the dropping funnel over a 10 min period, maintaining the internal temperature below 5 °C. The cooltetrahydrofuraning bath is removed and the solution is stirred at room temperature for 30 min. The dropping funnel is quickly replaced with a 50-mL dropping funnel and the solution of lithium t-butoxide is cooled with a sodium chloride/ice bath (internal temperature -15 °C). A solution of crude 5 (25.4 g) in dry tetrahydrofuran (15 mL) is added through the dropping funnel over a 10 min period, keeping the internal temperature below -10 °C. After rinsing the dropping funnel with dry tetrahydrofuran (5 mL), the reaction mixture is stirred below -10 °C for 20 min (Note 25). Saturated aqueous NH4Cl (100 mL) and water (100 mL) are added to quench the reaction and the mixture is transferred into a 1-L separatory funnel. The separated aqueous layer is extracted with diethyl ether (2 x 100 mL). The combined organic layers are washed with water (2 x 100 mL), brine (200 mL), and dried over anhydrous MgSO4, filtered through cotton, and then concentrated by rotary evaporation (70 mmHg, water bath temperature 40 °C) to afford crude carbamate 6, which is dried under vacuum (0.3 mmHg) to constant weight (21.15 g) (Note 26). The residual orange liquid is subjected to silica gel chromatography (Note 27). The combined fractions are concentrated by rotary evaporation (70 mmHg, water bath temperature 40 °C) and dried under vacuum (1.0 mmHg) (Note 8) to furnish Boc-carbamate 6 as a light-yellow oil (12.44 g, 69% overall yield from 1) (Note 28 and Note 29).
2. Notes
1. All glass apparatus was dried in an oven at 120 °C for 2 h and cooled in a drying desiccator.
2.
Geraniol (
1) (97%) was purchased from Wako Pure Chemical Industries, Ltd. and used directly without purification.
3. The submitters purchased
dichloromethane from Wako Pure Chemical Industries, Ltd. and dried over 4Å molecular sieves. The
water content determined by Karl-Fischer titration was <10 ppm. The molecular sieves were activated in a round-bottomed flask by heating in a household microwave oven (500 W) at full power for 5 min, and then cooled to room temperature under vacuum (1.8 mmHg) for 10 min. This procedure was repeated 3 times. The checkers purchased
dichloromethane from Kanto Chemicals Co., Inc. and purified by a Glass Contour Solvent Dispensing System. The
water content by Karl-Fischer titration was <10 ppm.
4.
Trichloroacetyl isocyanate (97%) was purchased from Wako Pure Chemical Industries, Ltd. and used as received.
5. The progress of the reaction was monitored by thin-layer chromatography (TLC) using E. Merck pre-coated 0.25 mm thick silica gel 60 F254 plates. The plates were eluted with v/v 3:1
n-hexane/ethyl acetate and visualized with 254 nm UV light followed by dipping in a
phosphomolybdic acid solution (w/v 3:20 in
ethanol) and heating on a hot plate. The product
trichloroacetyl carbamate 2 had an R
f = 0.58 and the starting material
geraniol (
1) an R
f = 0.33.
Trichloroacetyl carbamate 2 undergoes gradual hydrolysis to carbamate
3 on TLC resulting in formation of a new spot with an R
f = 0.29.
6. Methanol (99.8%) was purchased from Wako Pure Chemical Industries, Ltd. and used as received.
7. A slight temperature increase (ca 1-2 °C) was observed.
8. The submitters dried the material at 0.3 mmHg on a vacuum line.
9. After stirring at room temperature for 15 min, the solution became clear.
10. When using
ethyl acetate instead of
diethyl ether, the crude product contained some byproducts, which were detrimental in step B.
11. An analytically pure sample was obtained by dissolving the crude carbamate
3 (8.35 g) in
n-hexane (50 mL) and cooling in an ice bath for one hour. The precipitated white solid was quickly filtered and washed with chilled
n-hexane to provide pure carbamate
3 (4.63 g 55%).
(E)-3,7-Dimethylocta-2,6-dien-1-ylcarbamate(
3) exhibits the following properties: mp 30-31 °C; IR (NaCl): 3464, 3346, 2921, 1712, 1602 cm
-1;
1H NMR
pdf (400 MHz, CDCl
3) δ: 1.59 (s,3 H), 1.69 (s,3 H), 1.71 (s,3 H),
2.05-2.12 (m, 4 H), 4.56 (br s, 2 H), 4.59 (d,
J = 6.9 Hz, 2 H), 5.09 (br t,
J = 6.4 Hz, 1 H), 5.36 (br t,
J = 6.9 Hz, 1 H);
13C NMR
pdf (100 MHz, CDCl
3) d: 16.3, 17.6, 25.6, 26.2, 39.4, 61.8, 118.4, 123.6, 131.7, 142.0, 157.4; Anal. calcd for C
11H
19NO
2: C, 66.97; H, 9.71; N, 7.10. Found C, 66.90; H, 9.48; N, 7.10; HRMS (ESI):
m/z calcd for C
11H
20NO
2 (M+H
+ 198.1494, found 198.1495.
12.
Toluene (99.5%) was purchased from Wako Pure Chemical Industries, Ltd. and used as received.
13.
Triphenylphosphine (Extra Pure grade), purchased from Kanto Chemical Co., Inc., was dried in a desiccator over
phosphorus pentoxide under vacuum (ca. 20 mmHg) prior to use.
14.
Triethylamine, purchased from Wako Pure Chemical Industries, Ltd., was distilled from
CaH2 and stored over
potassium hydroxide pellets.
15.
Carbon tetrabromide (99%) was purchased from Tokyo Chemical Industry Co., Ltd. and used as received.
16. The reaction was monitored by TLC. Carbamate
3 has an R
f = 0.29 eluting with v/v 3:1
n-hexane/ethyl acetate and isocyanate
5 has an R
f = 0.40 (
n-hexane) and 0.80 (
n-hexane/diethyl ether 20:1).
17.
n-Hexane (95%) was purchased from Wako Pure Chemical Industries, Ltd. and used as received.
18. A porcelain Büchner funnel (4 cm diameter x 5 cm height) filled with 20 g of Celite was employed.
19. The submitters found that crude
5 can be purified by dissolution in
n-hexane followed by passing the solution through a short column of silica gel. After elution with
n-hexane followed by collecting and concentrating the fractions, isocyanate
5 was obtained as an oil, which was distilled using a Kugelrohr (0.83 mmHg, 50-55 °C). Although the resulting clear oil was homogeneous according to proton and carbon NMR spectroscopy, a correct elemental analysis could not be achieved. Spectral data for
3-isocyanato-3,7-dimethylocta-1,6-diene (
5)are as follows: IR (NaCl): 2977, 2931, 2257 cm
-1;
1H NMR
pdf (400 MHz, CDCl
3) d: 1.41 (s, 3 H), 1.56-1.64 (m, 2 H), 1.61 (s, 3 H), 1.68 (s, 3 H), 1.98-2.07 (m, 2 H), 5.05-5.08 (m, 1 H), 5.10 (d,
J = 10.6 Hz, 1 H), 5.27 (d,
J = 17.0 Hz, 1 H), 5.75 (dd,
J = 17.0, 10.5 Hz, 1 H);
13C NMR
pdf (100 MHz, CDCl
3) d: 17.6, 23.1, 25.6, 29.2, 42.8, 62.2, 112.8, 122.9, 123.1, 132.3, 141.9; HRMS (DART):
m/z calcd for C
11H
18NO (M+H)
+ 180.1388, found 180.1385.
20.
1H NMR analysis of the crude oil showed a small signal corresponding to the bromoform proton appearing at d 6.83. Bromoform does not affect the subsequent step.
21. The submitters purchased dry
tetrahydrofuran (anhydrous, 99+%) from Kanto Chemical Co., Inc. and used as received. The
water content was 1 ppm by Karl-Fischer titration. The checkers purchased dry
tetrahydrofuran from Kanto Chemicals Co., Inc., and purified by a Glass Contour Solvent Dispensing System. The
water content was 9.5 ppm by Karl-Fischer titration.
22.
t-Butyl alcohol, purchased from Wako Pure Chemical Industries, Ltd., was distilled from
CaH2 and stored over 4Å molecular sieves.
23.
Lithium hexamethyldisilazide (1 M solution in
tetrahydrofuran) was purchased from Sigma-Aldrich Chemical Company and used as received.
24. Initially, the submitters employed
lithium t-butoxide prepared by the reaction of
n-butyllithium with
t-butanol in
THF. In large-scale experiments, problems were sometimes encountered with the formation of a side product having a similar R
f value to that of Boc-carbamate
6. Further exploration showed that the side product was the
n-butyl carbamate as shown below.
Although this problem could be avoided by using freshly opened n-butyllithium coupled with rigorous degassing of t-butanol by freeze-thaw cycles immediately before use, lithium hexamethyldisilazide was ultimately selected as the base to obtain reproducible product purity and yields.
25. Reaction progress was monitored by TLC. The isocyanate starting material
5 has an R
f = 0.40 (
n-hexane) and the Boc-carbamate product
6 has an R
f = 0.47 (v/v 7:1
n-hexane/ethyl acetate).
26. It is important to remove bromoform at this stage because it coelutes with Boc-carbamate
6 during chromatographic purification.
27. The checkers purchased 'Silca Gel 60 (spherical)' (particle size 63-210 mm) from Kanto Chemical Co., Inc. The crude product was transferred to a column (8 x 30 cm, 100 g of silica gel) with a minimum amount of
n-hexane and eluted with 400 mL of
n-hexane. Elution was continued with 1.5 L of
n-hexane:
ethyl acetate (v/v 20:1) and fraction collection (100-mL) was begun. Fractions 4-10 were combined and concentrated by rotary evaporation.
28. The submitters reported purity of 97% by GC analysis using an Agilent 6890N instrument equipped with an Agilent J&W DB-5 column (30.0 m x 0.25 mm) and a flame ionization detector using a method of 160 °C isotherm for 10 min, then ramp 20 °C/min to 300 °C, then 300 °C isothermal for 8 min with flow rate 30 cm/sec He carrier gas. The retention time for the product was 6.9 min with unidentified small peaks having retention times 1.7-6.4 min.
29. Kugelrohr distillation of carbamate
6 (oven temperature 130-135 °C at 1.0 mmHg) afforded an analytically pure sample. Spectral data for
t-butyl 3,7-dimethylocta-1,6-dien-3-ylcarbamate(
6): IR (NaCl): 3277, 2975, 2929, 1717, 1698 cm
-1;
1H NMR
pdf (400 MHz, CDCl
3) δ: 1.37 (s,3 H), 1.43 (s,9 H), 1.60 (s,3 H), 1.65-1.69 (m, 2 H), 1.68 (s,3 H), 1.94 (q,
J = 7.3 Hz, 2 H), 4.61 (br s, 1 H), 5.05-5.12 (m, 3 H), 5.89 (dd,
J = 17.4, 10.5 Hz, 1 H);
13C NMR
pdf (100 MHz, CDCl
3) δ: 17.5, 22.5, 24.7, 25.6, 28.4, 39.5, 56.1, 78.8, 111.9, 124.0, 131.8, 143.5, 154.2; Anal. calcd for C
15H
27NO
2: C, 71.10; H, 10.74; N, 5.53. Found C, 70.71; H, 10.76; N, 5.53; HRMS (DART):
m/z calcd for C
15H
28NO
2 (M+H)
+ 254.2120, found 254.2117.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In the development and checking of these procedures, every effort has been made to identify and minimize potentially hazardous steps. The Editors believe that the procedures described in this article can be carried out with minimal risk if performed with the materials and equipment specified, and in careful accordance with the instructions provided. However, these procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Allyl cyanates generated under several reaction conditions are considered to be possible transient intermediates, which undergo instantaneous rearrangement below ambient temperature to afford allyl isocyanates.
2 Historically, the first attempts to synthesize allyl cyanates dates back to a report in 1970 by Holm, who employed a cheletropic reaction of allyl thiatriazoles and found that only rearranged allyl isocyanates were isolated.
3 In 1978, Overman examined the synthesis of allyl cyanates by using the reaction of
cyanogen chloride with the lithium salts of allyl alcohols and observed formation of a mixture of allylically rearranged isocyanates and dimeric carbamates.
4 Although these two pioneering reports support the belief that rearrangement of presumed allyl cyanates occurs below room temperature, development of an allyl cyanate-to-isocyanate rearrangement as a synthetic method for preparation of allyl amines is hampered by obstacles associated with preparation of cyanates (esters of cyanic acid: R-OCN).
5 Moreover, no mechanistic investigation had been undertaken to determine if the reaction proceeds via an ionic or concerted mechanism.
6 In 1991, Ichikawa introduced a new synthetic method for the preparation of allyl cyanates which involves
dehydration reactions of allyl carbamates.
7 This approach is quite reasonable considering the analogous methods to form carbon-
nitrogen triple bonds, such as dehydration reactions of amides or formamides to produce nitriles or isonitriles, respectively (Scheme 1).
8 Scheme 1. Retrosynthetic analysis of cyanates by analogy
Ichikawa examined several dehydration conditions of allyl carbamates and found that
trifluoromethanesulfonic anhydride (
Tf2O) and
diisopropylethylamine (
DIPEA) in
dichloromethane at -78 °C (method A)
9 and
triphenylphosphine,
carbon tetrabromide and
triethylamine (
TEA) in
dichloromethane at -20 °C (method B)
10 accomplish the transformation to form allyl cyanates (Scheme 2). It should be noted that these two dehydration methods were originally reported as procedures for the preparation of isonitriles from formamides. Although method B produces
triphenylphosphine oxide as co-product, which is often difficult to remove, we preferred method B over method A owing to its experimental simplicity. Furthermore, the reaction conditions employed in method B are sufficiently mild to tolerate a wide range of functional groups. One of the important features of this rearrangement reaction is that the allyl isocyanate products can be flexibly transformed to a variety of allyl amine derivatives. For example, addition of amines,
trimethylaluminum or alcohols to the reaction mixture containing the allyl isocyanate affords the respective ureas, acetamides
11 or carbamates.
12
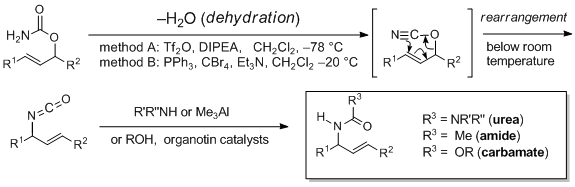
Scheme 2. Allyl cyanate-to-isocyanate rearrangement for the synthesis of allyl amine derivatives
The advantageous feature of the transformation of allyl isocyanates to allyl amine derivatives is demonstrated in synthetic studies of the nucleoside antibiotic,
blasticidin S (
11) (Scheme 3).
13 In the route to this target, transformation of
hex-3-enopyranose 7 to 4-amino-hex-2- enopyranose (
10a,
10b,
10c) was carried out by utilizing an allyl cyanate-to-isocyanate rearrangement (
8→9). Treatment of the produced allyl isocyanate
9 with
2,2,2-trichloroethanol,
benzyl alcohol or
allyl alcohol furnished the respective Troc (
10a), Cbz (
10b) and Alloc carbamate (
10c) derivatives. After considerable experimentation with these three carbamates to determine the best choice of protecting group, the Troc carbamate (
10a) was selected as a key intermediate, which led to the first total synthesis of
blasticidin S. In addition, it is noteworthy that the intermediate allyl cyanate
8, having the cyanate group at the pseudo-equatorial position, underwent smooth rearrangement even at 0 °C.
14
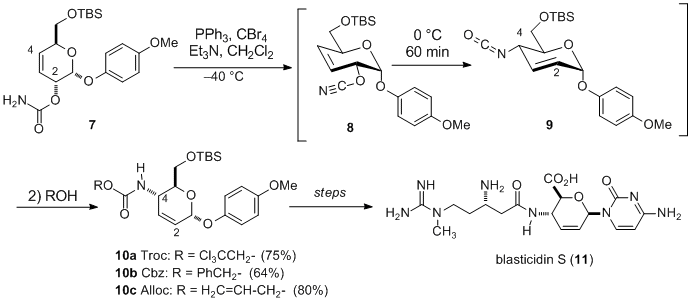
Scheme 3. The key step in the total synthesis of blasticidin S.
The stereochemistry of allyl cyanate-to-isocyanate rearrangement was examined using chiral allyl carbamate
13, which was prepared from
ethyl (S)-lactate (
12) (Scheme 4).
15 Dehydration of
13 followed by treatment of the resulting allyl isocyanate
15 with
trimethylaluminum furnished amide
16 in a good yield. The enantiomeric purity of the rearranged product
16 was determined to be 98% by analysis of the corresponding MTPA esters
17. The absolute stereochemistry of the formed stereogenic center in
16 was determined by analysis of the MTPA amides
18 using the Kusumi method for elucidation of the absolute configuration of primary amines.
16 The results of these studies led to the conclusion that the rearrangement of allyl cyanates proceeds via a concerted mechanism resulting in [1,3]-chirality transfer with a high level of stereoselectivity.
Scheme 4 Examination of stereochemistry in allyl cyanate-to-isocyanate rearrangement.
The availability of a reliable method for the preparation of allyl cyanates through dehydration of allyl carbamates coupled with the stereoselective nature of the rearrangement established that allyl cyanate-to-isocyanate rearrangement is a useful method for the enantioselective synthesis of allyl amine derivatives. As a result, a number of organic chemists have employed this reaction for the syntheses of
nitrogen-containing bioactive compounds.
17 The procedure described herein, highlighting the allyl cyanate-to-isocyanate rearrangement, serves as an efficient method for the preparation of allyl amines as Boc-carbamate derivatives starting from allyl alcohols. This is exemplified by conversion of
geraniol (
1) to the Boc-carbamate
6. Carbamoylation of
1 is carried out by treatment with
trichloroacetyl isocyanate followed by hydrolysis of the trichloroacetyl group of
2 with
potassium carbonate in aqueous
methanol to provide allyl carbamate
3 in excellent yield (Step A).
18 Although
trichloroacetyl isocyanate is an expensive and moisture-sensitive liquid that causes difficulties in handling, it is an ideal reagent for this transformation due to its high reactivity, clean reaction, and excellent yields.
19 Dehydration of allyl carbamate
3 generates allyl cyanate
4, which undergoes spontaneous rearrangement to afford allyl isocyanate
5 (Step B). Although the dehydration conditions are mild enough to tolerate a wide range of functional groups, one drawback is the formation of the co-product
triphenylphosphine oxide. In the present case, dissolution of the crude reaction mixture in
hexane results in precipitation of
triphenylphosphine oxide, which is easily removed by filtration. In smaller scale experiments, it is more convenient to purify the product directly by chromatographing the crude mixture rather than by precipitation and filtration. When the product has the same or similar R
f value as
triphenylphosphine oxide,
tributylphosphine can be used as an alternative.
12 Other dehydration methods, which circumvent problems associated with
triphenylphosphine oxide, utilize either Tf
2O/DIPEA
7 or trifluoroacetic anhydride (TFAA)/TEA.
20 In particular, in their total synthesis of manzamine A, Fukuyama and co-workers employed TFAA/TEA for dehydrating the functional group-rich carbamate
19 (Scheme 5).
21 It should be noted that use of the TFAA/TEA procedure was originally described as a synthetic method for the preparation of nitriles from amides by Casini.
22
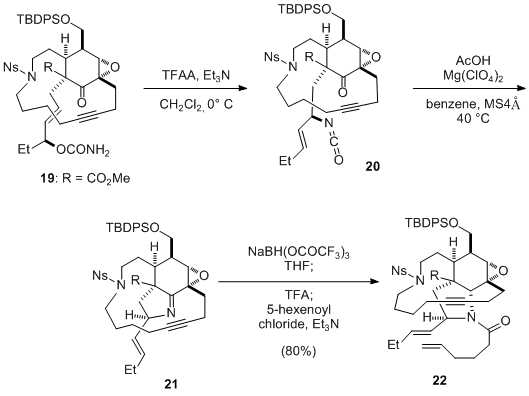
Scheme 5 The key step in the total synthesis of manzamine A
Transformation of the sterically crowded isocyanate
5 to the Boc-carbamate
6 was performed by reaction with
lithium t-butoxide. Since
lithium t-butoxide is a strong base, we cannot employ these conditions with isocyanates containing base-sensitive functional groups or epimerization-prone stereogenic centers. In these cases, it is recommended that related procedures be used to transform hindered isocyanates into carbamates under Lewis or Brønsted acid conditions using
titanium tetra-t-butoxide23 or
trimethylsilyl chloride24 or
molybdenum (VI) dichloride dioxide.
25 In summary, the transformation of
geraniol (
1) to Boc-carbamate
6 exemplifies a useful synthetic method for the construction of quaternary carbons bearing
nitrogen substituents.
26
Appendix
Chemical Abstracts Nomenclature; (Registry Number)
(E)-3,7-Dimethylocta-2,6-dien-1-ylcarbamate (3) (16930-44-2)
Geraniol (1) (106-24-1)
Trichloroacetyl isocyanate (3019-71-4)
tert-Butyl 3,7-dimethylocta-1,6-dien-3-ylcarbamate (6) (1354913-05-5)
Triphenylphosphine (603-35-0)
Triethylamine (121-44-8)
Carbon tetrabromide (558-13-4)
Lithium hexamethyldisilazide (4039-32-1)

|
Yoshiyasu Ichikawa was born in Gamagori, Aichi in 1958. He completed his undergraduate studies and PhD at Nagoya University. After postdoctoral studies in the Dyson Perrins Laboratory at Oxford, UK he joined the faculty of Mie University where he pursued his interests in the synthesis of marine natural products based upon the sigmatropic rearrangements. He moved to Nagoya University in 1992 and emigrated to Kochi University in 2004. His research interests are in the area of synthesis of natural products and carbohydrate chemistry.
|

|
Noriko Kariya was born in 1986 in Kochi, Japan. She carried out her undergraduate research with Professor Yoshiyasu Ichikawa at Kochi University where she received her MS degree in 2012.
|

|
Tomoyuki Hasegawa was born in 1988 in Shizuoka, Japan. He pursued his undergraduate studies with Professor Yoshiyasu Ichikawa at Kochi University where he received his MS degree in 2013.
|

|
Eiji Yoshida was born in Ann Arbor, Michigan in 1989. He received his B.S. in 2012 from the University of Tokyo. In the same year, he began his graduate studies at the Graduate School of Pharmaceutical Sciences, the University of Tokyo, under the guidance of Professor Tohru Fukuyama. His research interests are in the area of the total synthesis of natural products.
|
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved