1. Procedure
A. 4-Phenyl-1,2-butadiene (1). A 3-necked 250-mL round-bottomed flask containing a magnetic stir bar (3 cm length x 1.5 cm diameter) is equipped with a reflux condenser and two rubber septa (Note 1). The top of the condenser is fitted with a stopcock connected to a vacuum line with an argon flow. Magnesium turnings (7.2 g, 0.3 mol, 1.2 equiv) are added and the apparatus is flame-dried under vacuum. After cooling to room temperature under an argon purge, the flask is charged with diethyl ether (100 mL) and equipped with a thermometer. 1,2-Dibromoethane (0.5 mL) (Note 2) is added to initiate the reaction with a slight increase of the internal temperature (2-3 °C). Benzyl bromide (30 mL, 0.25 mol) (Note 2) is injected into the mixture with a syringe pump at a rate so as to maintain a gentle reflux (15 mL/h). After the addition, the mixture is heated under reflux with an oil bath (oil bath temperature, 55 °C) for another 2 h. Another 3-necked 250-mL round-bottomed flask equipped with a magnetic stir bar (3 cm length x 1.5 cm diameter) (Note 1) is fitted with two rubber stoppers and a 250-mL pressure equalizing addition funnel. The flask and funnel are flame-dried under vacuum. After cooling to room temperature under an argon purge, propargyl bromide (33.4 mL of an 80 wt. % solution in toluene, 0.3 mol, 1.2 equiv) (Note 2) and diethyl ether (50 mL) (Note 2) are added and the flask is fitted with a thermometer. The mixture is cooled to 0 °C (internal temperature) with an ice-salt bath. The prepared Grignard reagent is transferred to the addition funnel using an 18-gauge cannula and added to the propargyl bromide over 3 h. After the addition, the resulting mixture is stirred for an additional 2 h at 0 °C (Note 3). Then the mixture is poured into a 1-L Erlenmeyer flask containing a cold saturated aqueous solution of NH4Cl (200 mL). The mixture is transferred to a 500 mL separatory funnel, the phases are separated, and the aqueous phase is back-extracted with diethyl ether (100 mL x 2) (Note 2). The combined organic phases are washed with brine (100 mL), dried over anhydrous Na2SO4, and filtered. The solvent is removed by rotary evaporation (20 mmHg, 30 °C water bath). Purification is performed by vacuum distillation (70-80 °C/23 mmHg, fractionating column: 2 cm diameter x 15 cm length) to afford 4-phenyl-1,2-butadiene(1) (18.6 g, 57% by the checker; 24.9 g, 77% by the submitter, see discussion) as a colorless liquid (Note 4).
B.
1-Acetyl-2-phenethylidenecyclopropanecarboxylic acid ethyl ester (2). A 3-necked 250-mL round-bottomed flask is equipped with a reflux condenser, the top of which is fitted with a stopcock connected to a vacuum line with an
argon flow. The other two necks are fitted with rubber septa. After evacuating and backfilling with
argon the flask is charged with a magnetic stir bar (3 cm length x 1.5 cm diameter), Rh
2(OAc)
4 (180 mg, 0.4 mmol, 0.008 equiv) (
Note 5),
4-phenyl-1,2-butadiene (
1) (19.5 g, 150 mmol, 3.00 equiv) (
Note 6) and
CH2Cl2 (40 mL) (
Note 2) under
argon. The mixture is heated to reflux using an oil bath (55
°C, external temperature) and after 5 min at reflux, a solution of
ethyl α-diazoacetoacetate2 (7.8 g, 50 mmol, 1 equiv) in
CH2Cl2 (10 mL) is added using a syringe pump (10 mL/h). Upon completion of the addition, an additional quantity of
Rh2(OAc)4 (40 mg, 0.1 mmol, 0.002 equiv) (
Note 7) is added and the resulting mixture is refluxed for 2 h; the progress of the reaction is monitored by TLC (
Note 3). Upon completion, the reaction is cooled to room temperature and concentrated by rotary evaporation (20 mmHg, 35 °C water bath). The residue is purified by column chromatography (column ∅ = 80 mm with 300 g of silica gel (230-400 mesh); ~150 mL fractions;
n-hexane (1.5 L) (
Note 2) is used to recover
4-phenyl-1,2-butadiene 1 (fractions 5-12, 7.64 g, 39%); then a mixture of 3:1
n-hexane:ethyl acetate (
Note 2) (2 L) is used to elute 1-acetyl-2-phenethylidene- cyclopropanecarboxylic acid ethyl ester
2 (fractions 20-25, 15.1 g ) (
Note 8).
2. Notes
1. All glassware was thoroughly washed and dried in an oven (150 °C). Magnetic stir bars were washed with
acetone and dried.
2. The submitters prepared their own
magnesium turnings from
magnesium ingot (>99%) purchased from Sinopharm Chemical Reagent Co., Ltd and obtained a higher yield for the reaction (77% vs 57%). The checkers used
magnesium turnings purchased from Sigma Aldrich. The submitters used anhydrous
diethyl ether (=99%) purchased from Shanghai Experimental Reagent Co., Ltd and distilled over sodium wire with
diphenyl ketone as the indicator and the checkers used column-dried
diethyl ether. The submitters used
dichloromethane distilled from
calcium hydride and the checkers used column-dried
dichloromethane.
Benzyl bromide (=98%) was purchased from Sigma Aldrich and distilled from
MgSO4 before use. The submitters purchased
propargyl bromide (=98%) from Shanghai Darui Finechemical Co., Ltd and distilled before use. The checkers purchased an 80 wt. % solution of
propargyl bromide in
toluene (Sigma-Aldrich) and used as received.
1,2-Dibromoethane (=98%) was purchased from Sigma-Aldrich, petroleum ether from Fisher Scientific, and all the other chemicals from Sigma Aldrich. All chemicals were used as received unless specified above.
Ethyl diazoacetoacetate was prepared using an
Organic Syntheses method.
2
3. TLC analysis: Step A: R
f of compound
1 = 0.68 (eluent: petroleum ether); Step B: R
f of
ethyl α-diazoacetoacetate = 0.39 (eluent: petroleum ether/EtOAc (10/1)) and visualized using UV (254 nm); R
f of compound
2 = 0.42 (eluent: petroleum ether/EtOAc (10/1)) and visualized with KMnO
4.Step C: R
f of compound
3 = 0.48 (eluent: petroleum ether/EtOAc (10/1)) and visualized using UV (254 nm) and an aqueous solution of KMnO
4.
4.
4-Phenyl-1,2-butadiene 6 was colorless after distillation under reduced pressure, but turned yellow after a few hours at room temperature. GC purity: 94.6% (conditions: Rtx-5MS column (30x.25x.25); oven: 50 °C, then 10
°C/min, 270 °C for 5 min; injector: 230 °C; EI, interphase temperature: 280 °C; split: 42:1; He: 3.0 mL/min).
1H NMR
pdf(400 MHz, CDCl
3) δ: 3.36x3.40 (m, 2 H), 4.72-4.75 (m, 2 H), 5.30 (quint,
J = 7.0 Hz, 1 H), 7.21-7.34 (m, 5 H);
13C NMR
pdf(100 MHz, CDCl
3) δ: 35.3, 75.2, 89.7, 126.4, 128.5, 128.6, 140.4, 209.1; IR (neat) n 3085, 3062, 3027, 2978, 2909, 2859, 1936, 1686, 1603, 1519, 1496, 1427, 1336, 1271, 1180, 1085, 1030, 968 cm
-1; MS (EI)
m/z 131 (89, M+H
+), 103 (100), 91 (98), 77 (88), 65 (80).
5. The submitters prepared
rhodium(II) acetate dimer (
Rh2(OAc)4) according to the procedure in
Inorganic Synthesis.
3 The checkers purchased
Rh2(OAc)4 from Strem Chemicals.
6. The submitters reported an increase in the yield of this reaction with increasing quantities of
4-phenyl-1,2-butadiene: 2.0 equiv gave 41% yield; 3.0 equiv gave 53% yield, and 4 equiv gave 60% yield of
2. After the reaction, excess
4-phenyl-1,2-butadiene was recovered by column chromatography.
Rh2(OAc)4 should be added in two portions because
ethyl α-diazoacetoacetate did not fully react when the same quantity of
Rh2(OAc)4 was added in one portion.
8. The crude
1-acetyl-2-phenethylidenecyclopropanecarboxylic acid ethyl ester 2 (a mixture of
Z/E isomers,
9 ratio = 1.7:1) was used directly in the next step without further purification. A portion of this crude material (60.8 mg) was purified by chromatographic separation on silica gel with an eluent of
n-hexane:ethyl acetate = 30:1 for characterization data.
1H NMR
pdf(300 MHz, CDCl
3) δ: 1.23-1.28 (m, 3 H), 2.07-2.32 (m, 2 H), 2.32-2.35 (m, 3 H), 3.50-3.56 (m, 2 H), 4.15-4.23 (m, 2 H), 6.01 (t,
J = 7.2 Hz, 0.34 H, =CH), 6.05-6.12 (m, 0.66 H, =CH)], 7.13-7.31 (m, 5 H);
13C NMR
pdf(100 MHz, CDCl
3) δ: 14.2 (2 C), 17.7, 19.0, 28.5, 28.7, 37.5, 37.9, 39.9 (2 C), 61.6 (2 C), 118.6 (2 C), 125.1, 125.2, 126.5 (2 C), 128.6 (2 C), 128.7 (2 C), 139.3, 139.4, 169.0, 169.1, 200.4, 200.8; IR (neat) n 1686, 1496, 1427, 1356, 1273, 1235, 1207, 1180, 1129, 1084, 1030 cm
-1; MS
m/z 259 (M+H
+, 100), 258 (39), 213 (13), 212 (7); HRMS calcd. for C
16H
19O
3: 259.1334. Found: 259.1332.
9.
1-Benzyl-2-methyl-3-(ethoxycarbonyl)-4-(2-phenylethyl)-1H- pyrrole (3): mp = 42 °C;
1H NMR
pdf(400 MHz, CDCl
3) δ: 1.37 (t,
J = 7.1 Hz, 3 H), 2.44 (s, 3 H), 2.88-2.92 (m, 2 H), 2.99-3.03 (m, 2 H), 4.32 (q,
J = 7.1 Hz, 2 H), 4.97 (s, 2 H), 6.30 (s, 1 H), 6.96 (d,
J = 6.7 Hz, 2 H), 7.16-7.34 (m, 8 H);
13C NMR
pdf(100 MHz, CDCl
3)
δ: 11.8, 14.7, 29.1, 37.2, 50.4, 59.3, 111.3, 119.7, 124.9, 125.7, 126.5, 127.7, 128.2, 128.7, 128.9, 136.6, 137.3, 142.8, 166.2; MS (EI)
m/z 348 (M+H
+, 100), 347 (4), 302 (2), 256 (3); IR (neat) n 1950, 1686, 1642, 1564, 1506, 1496, 1445, 1428, 1355, 1339, 1273, 1235, 1180, 1129, 1084, 1030 cm
-1; HRMS (EI) calcd for C
23H
26NO
2+ (M
+): 348.1964; Found: 348.1954; Anal. calcd. for C
23H
25NO
2: C, 79.51; H, 7.25; N, 4.03; Found: C, 79.67; H, 7.15; N, 3.89.
10.This compound was recrystallized from petroleum ether and
EtOAc for melting point determination. For recrystallization,
3 (0.53 g) was dissolved in
EtOAc (1 mL) with heating. Then petroleum ether (20 mL) was added. The solution was sealed and kept at -8 °C for one day. The crystals (65 mg, 12%) were collected via filtration and 465 mg (88%) was recovered by evaporation of the mother liquor.
3. Discussion
Pyrroles are one of the most prevalent heterocyclic compounds. In recent years, polysubstituted pyrroles with various substituents have been prepared.
4 However, there are very limited reports on syntheses of 2,3,4-trisubstituted pyrroles.
5 In this contribution, we have demonstrated an intermolecular cyclization reaction of alkylidenecyclopropyl ketones and amines which provides an efficient route to 2,3,4-trisubstituted pyrroles.
4-Phenyl-1,2-butadiene was first prepared by the group of Hirao et al. by first generating an organovanadium compound from reaction of
benzylmagnesium bromide with VCl
3, then reacting the resulting species with
propargyl bromide to afford the
allene in 31% yield with 4% of
4-phenyl-1-butyne as a byproduct.
6 Other methods of generating alkyl- and aryl-substituted allenes have been mediated by CuX.
7 However, we found
4-phenyl-1,2-butadiene could be prepared without CuX or any additional metal as catalyst. New glassware was used to ensure no residual Cu(I) was present as a contaminant. Initial efforts using this procedure resulted in the formation of >10% of
4-phenyl-1-butyne.
7 However, its formation was diminished by slow addition of the Grignard reagent into
propargyl bromide. The preparation of
benzyl magnesium bromide is very important to ensure a higher yield. The checkers did not see the formation of
4-phenyl-1-butyne but instead observed a 16% recovery of
propargyl bromide and an 11% yield of
1,2-diphenylethane as estimated by crude NMR, due to the less efficient formation of the
benzyl magnesium bromide forming the
benzyl bromide-homocoupling product
1,2-diphenylethane leading to the recovery of the
propargyl bromide in the subsequent step.
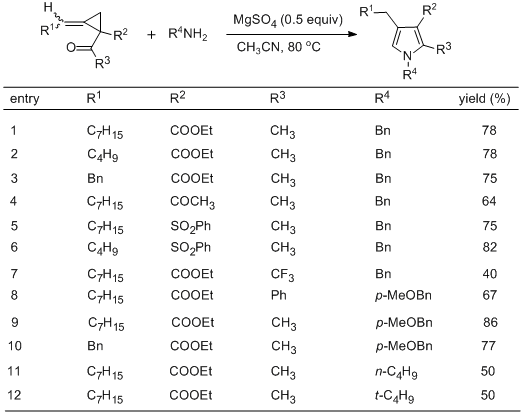
We have prepared various 2,3,4-trisubstituted pyrroles via reaction of alkylidenecyclopropyl ketones with amines (Table 1).
8 In step B, the alkylidenecyclopropylketone products are contaminated with some minor by-products, but the crude product obtained by simple filtration through a column of silica gel can be used directly for the next step. In the multi-gram scale reaction of step C, moisture is easily controlled; thus, the reaction can be conducted without
MgSO4.
Table 1. Intermolecular Cyclization of Alkylidenecyclopropyl Ketones with Amines Affording 2,3,4-Trisubstitued Pyrroles
Two possible mechanistic pathways can be envisioned to afford the corresponding pyrroles. One pathway starts from nucleophilic attack of the amine at the less sterically hindered carbon atom of the 3-membered ring to afford intermediate
4 (path a). The nucleophilic nitrogen then reacts with the carbonyl group leading to 3-alkylidene-5-hydroxy tetrahydropyrrole intermediate
5. Subsequent dehydration and aromatization generates product
3 via the intermediacy of
6. An alternative pathway starts with the intermolecular condensation of
1 and
2 to afford the cyclopropylimine intermediate
7 (path b).Cloke-type rearrangement of intermediate
7 readily leads to ring expansion via the subsequent nucleophilic attack of the nitrogen atom at the less sterically hindered carbon atom in the cyclopropane ring, which causes the distal cleavage to form
6.
10 Subsequent aromatization affords product
3.
Scheme 1. Proposed reaction mechanism
Appendix
Chemical Abstracts Nomenclature; (Registry Number)
Magnesium (7439-95-4)
Benzene, (bromomethyl)- (100-39-0)
1-Propyne, 3-bromo- (106-96-7)
Magnesium, bromo(phenylmethyl)- (1589-82-8)
Benzenemethanamine (100-46-9)
Butanoic acid, 2-diazo-3-oxo-, ethyl ester (2009-97-4)
Rhodium, tetrakis[µ-(acetato-κO:κO')]di-, (Rh-Rh) (15956-28-2)
Ethane, 1,2-dibromo- (106-93-4)
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved