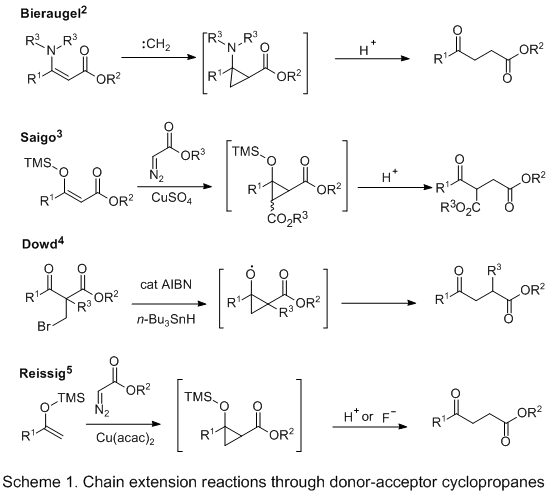
Methods for the preparation of donor-acceptor (push-pull) cyclopropanes for the purpose of incorporating a single carbon between two carbonyl groups have been developed by a number of research groups
(Scheme 1). Bieräugel described the cyclopropanation of a β-keto ester-derived enamine, which upon hydrolysis provided homologated material.
2 Attempts by Saigo to mimic these results with silyl enol ethers were inefficient and provided mixtures of products when using zinc carbenoids, although reactions based on copper-carbenoids derived from diazoesters were more efficient.
3 A radical-based method for homologation of β-dicarbonyls was reported by Dowd, although the one-carbon insertion was limited to α-substituted β-dicarbonyl starting materials.
4 A complementary strategy for the formation of donor-acceptor cyclopropanes was reported by Reissig, in which methyl ketone-derived enol ethers were reacted with stabilized rhodium carbenoids.
5 The resulting cyclopropanes could be converted cleanly to γ-keto esters through hydrolysis and used as nucleophilic species in tandem reaction processes.
A one-pot zinc carbenoid-mediated homologation reaction was reported by Zercher and co-workers.
6 Treatment of unfunctionalized β-keto esters with the Furukawα-modified Simmons-Smith reagent,
7 generated from an equimolar ratio of
diethylzinc and
diiodomethane, provided rapid and efficient access to γ-keto esters
(Scheme 2). Labeling studies revealed that the carbenoid carbon was inserted regioselectively adjacent to the ketone functionality, an observation that suggested the intermediacy of a donor-accepter cyclopropane.
Mechanistic understanding of the zinc carbenoid-mediated homologation reaction was also informed by computational investigations, NMR analyses of reactive intermediates, and studies involving reaction stoichiometry.
8 The proposed reaction mechanism is summarized in
Scheme 3. After initial deprotonation by zinc carbenoid (or
diethylzinc), the resulting enolate reacts with carbenoid to provide homoenolate
3. Intramolecular cyclization into the more electrophilic carbonyl provides the donor-acceptor cyclopropane (
4), which fragments with a low energy barrier to provide an organometallic intermediate. This species is structurally reminiscent of the traditional zinc-Reformatsky intermediate,
8 and the strong covalent character of the carbon zinc bond is likely responsible for the absence of its reactivity with the various alkylating electrophiles (carbenoid and ethyl iodine) present in solution.
The proposed mechanism illustrates that two equivalents of
diethylzinc are necessary to effect the transformation, although in practice three equivalents of
diethylzinc are used to ensure that the reactions proceed to completion. If acidic protons are present in the reaction substrate, the use of additional
diethylzinc might be necessary and does not hinder the homologation reaction. Neat
diethylzinc usually offers superior results, although commercial solutions of
diethylzinc can be used to avoid the handling of the pyrophoric neat reagent. The replacement of
diethylzinc with an alternate base for the purpose of enolate formation, thereby reducing the amount of pyrophoric reagent required in the reaction, should only be undertaken with the understanding that the stability of the zinc-organometallic intermediate
5 may be compromised in the presence of an alternate counterion. Reactions performed in the presence of non-zinc counterions result in greater product diversity and lower isolated yields of the simple homologated product.
10
The zinc carbenoid-mediated transformation described herein offers a number of advantages to many of the other donor-acceptor cyclopropane methods. For instance, easily accessible and often commercially available β-keto carboxylic acid derivatives serve as starting materials with no need for derivatization as enol ethers or enamines. Furthermore, no hydrolysis step is necessary to fragment the cyclopropane, which means that protic quenching of the reactive intermediate can be delayed until after homologation is complete and tandem reactions are performed. The zinc carbenoid-mediated homologation reaction operates on a wide variety of β-keto carboxylic acid derivatives,
11, 12and β-keto phosphonates
13 can also be transformed into the γ-keto homologues
(Scheme 4).
α-Substitution present on β-keto carboxylate starting materials often results in poor yields of chain extended or ring expanded products due to further reaction of the intermediate enolate; in contrast, α-substitution on β-keto phosphonate starting materials is well tolerated. Treatment of α-carboxyester cycloketones with carbenoid provides modest yields of the ring-expanded products, with use of more electrophilic carbenoids and control of stoichiometry being key to maximizing the efficiency of the transformation
(Scheme 5).14
Substituted carbenoids provides the means to incorporate functionality at the β-position of the γ-keto ester products. For example, the carbenoid prepared from 1,1-diiodoethane leads to regioselective incorporation of a methyl group adjacent to the ketone functionality
(Scheme 6) while preserving enolate character developed adjacent to the ester.
15
The utility of the zinc-carbenoid-mediated homologation reactions has been enhanced through its application to tandem reaction protocols. The zinc enolate, which is regioselectively incorporated adjacent to the ester functionality, can react with electrophiles to effect the tandem reactions. While most of the tandem sequences described below are applicable to the array of β-keto carboxylic acid derivatives, β-keto phosphonate substrates are often poor partners in the tandem reaction processes. While a variety of electrophiles can be used to effect tandem reactions, not all electrophiles react efficiently with the intermediate. For example, alkylation of the organometallic intermediate (enolate) is slow, which is consistent with its similarity to the traditional zinc-Reformatsky intermediate.
8 Therefore, incorporation of alkyl substituents at the α-carbon of the homologated material must rely upon indirect methods, some of which are highlighted below. Fortunately, many electrophiles react efficiently with the organometallic intermediate to provide access to a diverse array of α-substituted γ-keto esters.
Exposure of the organometallic intermediate
5 to aldehydes or ketones provide tandem aldol products.
16 Use of ester or amide starting materials provides selective access to
syn-aldol products with diastereocontrol as high as 20:1
(Scheme 7a). The syn selectivity is attributed to the influence of the γ-keto functionality, which is believed to favor formation of an intermediate Z-enolate.
17 Reaction through a closed transition state would then account for the
syn-selectivity. Tandem homologation-aldol reactions on substrates that possess an Evans auxiliary result in formation of
anti-aldol products with excellent enolate facial selectivity
(Scheme 7b). The anti-selectivity is consistent with the stereochemistry observed when excess Lewis acid is used in the reaction of oxazolidinone-derived Z-enolates.
18 Due to the use of three or more equivalents of
diethylzinc in the homologation portion of the reaction, excess zinc(II) is likely responsible for the aldol reaction proceeding through an open transition state.
The products of tandem homologation aldol reactions exist in hemi-acetal forms, which are attractive precursors to substituted tetrahydrofuranyl systems. A tandem homologation-aldol reaction, which utilized a carbenoid-derived from 1,1-diiodoethane, provided rapid access to the trisubstituted ring system, which was eventually converted to a
cis, cis-phaseolinic acid derivative via oxidative cleavage
(Scheme 8). The presence of the methyl group adjacent to the enolate was responsible for controlling both enolate facial and the
anti-aldol selectivities.
19 The aldol products can also be oxidized to provide 1,4-diketones, which have been used as precursors for heterocycle formation.
20
The organometallic intermediate can also be trapped with imine functionality, which provides a rapid route to substituted β-proline derivatives.
21 While the yields are modest and the chiral sulfonamide-derived imines cannot be used due to carbenoid reactivity with the sulfur, diastereocontrol can be effected through selection of the nitrogen activating group. Deprotection and reduction of the cyclic imine completes the preparation of β-proline derivatives
(Scheme 9). Enantiomerically-enriched products can be accessed through the use of oxazolidinone auxiliaries.
The stability of the intermediate organometallic species (
5) generated from a β-keto ester is demonstrated through the absence of its reaction with excess carbenoid, even over an extended period of time; however, the addition of catalytic
trimethylsilyl chloride to the organometallic intermediate induces reaction with carbenoid to effect formation of an intermediate ester homoenolate.
22 Protonation completes the installation of a methyl group, while formation of an iodomethyl side chain through treatment with iodine
23 provides access to a versatile functional group for further manipulation
(Scheme 10).
The use of the electrophilic carbenoid for carbon-carbon bond formation, therefore, provides a route to products unavailable from direct alkylation with alkyl halides.
Direct treatment of the organometallic intermediate with iodine provides access to the isolable α-iodo-species,
17 which can be efficiently transformed to an a,β-unsaturated-γ-keto ester functionality by treatment with base.
24 This transformation has been a key step in the synthesis of numerous natural products, including (+)-brefeldin A
25 (Scheme 11) and patulolides A and B.
26 In addition, carbon nucleophiles can be incorporated into these products via regioselective conjugate addition, which provides another indirect route to the incorporation of an alkyl group at the α-carbon.
27
Exposure of the organometallic intermediate to anhydrides is effective for the production of α-acylated products. The products produced from tandem
homologation-acylation reactions are β-keto esters, which are appropriate substrates for heterocycle formation through Paul-Knorr reactions.
20
In addition, the acylated products are appropriate β-keto ester substrates for which a second homologation reaction would effect formation of double chain extended products. One example of a tandem chain extension-acylation reaction involves treatment of methyl acetoacetate with carbenoid, followed by addition of methoxymaleic anhydride, which generates an acylated product that spontaneously cyclizes to generate a spiroketal. Members of the papyracillic acid family of natural products were successfully prepared through the use of this strategy
(Scheme 12).28
The reactions of β-diketones differ significantly from the reactions of β-keto esters, amides, or phosphonates. While the installation of the carbenoid carbon between the two ketone groups is believed to proceed through fragmentation of a donor-acceptor cyclopropane, the resulting intermediate is a reactive enolate as opposed to the stable organometallic formed from β-keto esters. The enolate reacts with additional equivalents of the carbenoid to form chain-extended cyclopropanol products
(Scheme 13).
6, 29 The absence of similar cyclopropanol products when starting with β-keto esters reflects the differences between zinc enolates of ketones and zinc enolates of carboxylic acid derivatives.
Strongly electrophilic carboxylic acid derivatives, such as acylated oxazolidinones, also provide access to cyclopropanated products when exposed to carbenoid for extended reaction times.
12, 23
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved